Jul 22, 2024
Viral genome analysis using CLC Genomics Workbench
- 1Yamagata prefectural institute of public health



External link: https://doi.org/10.7883/yoken.jjid.2025.055
Protocol Citation: Kenichi Komabayashi 2024. Viral genome analysis using CLC Genomics Workbench. protocols.io https://dx.doi.org/10.17504/protocols.io.kxygx3dnzg8j/v1
Manuscript citation:
Kenichi Komabayashi, Shuji Chikaoka, Hiroki Awano, Akiko Abe, Dai Sendo, Masayuki Furuyama, Masatoshi Kitamura, Saori Tanabe, Manabu Wada, Naomi Ogawa, Mika Sasaki, Yoko Aoki, Yohei Matoba, Junji Seto, Tatsuya Ikeda, Katsumi Mizuta (2025) Parechovirus A3 infections, including myalgia, in Yamagata, Japan between 2022 and 2023 and their next-generation sequencing analyses.Japanese Journal of Infectious Diseases doi: 10.7883/yoken.jjid.2025.055
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: February 05, 2024
Last Modified: July 22, 2024
Protocol Integer ID: 94708
Keywords: Genome Sequencing, metagenome, virus, bioinformatics, NGS, viral genome analysis, viral genome sequence, complete viral genomic sequence, complete viral genomic sequence from illumina, length viral genome sequence, clc genomic workbench, clc genomics workbench this protocol, using clc genomics workbench, genomic workbench, sequencing read, genome sequence, workflow of virustap, viral contig, virustap, genomic, genome, de novo assembly, sequence, blast program, length sequence, raw read, consensus sequence, clc, sequencing
Disclaimer
DISCLAIMER – FOR INFORMATIONAL PURPOSES ONLY; USE AT YOUR OWN RISK
The protocol content here is for informational purposes only and does not constitute legal, medical, clinical, or safety advice, or otherwise; content added to protocols.io is not peer reviewed and may not have undergone a formal approval of any kind. Information presented in this protocol should not substitute for independent professional judgment, advice, diagnosis, or treatment. Any action you take or refrain from taking using or relying upon the information presented here is strictly at your own risk. You agree that neither the Company nor any of the authors, contributors, administrators, or anyone else associated with protocols.io, can be held responsible for your use of the information contained in or linked to this protocol or any of our Sites/Apps and Services.
Abstract
This protocol aims to determine an nearly complete viral genomic sequence from Illumina sequencing reads which include those originating from virus, host cell, and sometimes bacteria. In the primary analysis, the raw reads undergo a filtration process to remove non-viral sequences, followed by de novo assembly to construct viral contigs. Subsequently, BLAST program is employed to identify the full-length viral genome sequence which is publicly available and shows high homology to the contig. In the secondary analysis, the full-length sequence is used as a reference to align Illumina sequencing reads and to generate a consensus sequence, resulting in a nearly complete viral genomic sequence.
We referred the workflow of VirusTAP by Yamashita et al. to built our analysis workflow method using the CLC genomic workbench.
Guidelines
This protocol is intended to analyze Illumina sequencing data to obtain nearly full-length viral genome sequences. The data to be input into the workflow are assumed to include sequences derived from hosts and bacteria as well as viruses, but non-viral sequences need to be reduced. For example, sequencing is performed after pretreatment with filters and nucleases, as in the method shown at dx.doi.org/10.17504/protocols.io.3byl4qnqzvo5/v1.
There are two advantages of the protocol.
1, Theoretically, the protocol can be adapted to any virus for which full-length genome sequence data are publicly available, regardless of species.
2, Data derived from multiple viral species or data from viruses with segmented genomes, such as influenza viruses, are also applicable.
Materials
Software
CLC Genomics Workbench
NAME
windows 11
OS
Qiagen
DEVELOPER
Software
CLC Microbial Genomics Module
NAME
Windows 11
OS
Qiagen
DEVELOPER
Software
CLC Genome Finishing Module
NAME
Windows 11
OS
Qiagen
DEVELOPER
Input data:
Paired end short read data (.fastq) which were demultiplexed prior to analysis
Preparation of data
Software
CLC Genomics Workbench
NAME
windows 11
OS
Qiagen
DEVELOPER
Hereafter abbreviated as CGW.
Software
CLC Microbial Genomics Module
NAME
Windows 11
OS
Qiagen
DEVELOPER
Hereafter abbreviated as CMGM.
CMGM is a plug-in for CGW, designed for the analysis of microbial genomes.
Software
CLC Genome Finishing Module
NAME
Windows 11
OS
Qiagen
DEVELOPER
Hereafter abbreviated as CGFM.
CGFM is a plug-in for CGW, designed to help finishing small genomes such as bacterial genomes.
Preparation of the host genome data.
Investigate the cell line utilized for virus cultivation.
Determine the taxonomic name of the organism from which the cell line originates.
To acquire reference genome sequences, access the 'References' in CGW or visit the NCBI database.
If the host cells originate from humans, one can utilize human genome data like hg38, which is prepared by QIAGEN. For further details, refer to the CGW manual 'QIAGEN Sets' (chapter 9.2 for ver. 23).
The University of California Genome Browser Gateway is a valuable resource for accessing reference genome sequences of various host organisms.
To create taxonomic profiling index from a reference database, use 'Create Taxonomic Profiling Index tool'.
Refer to the CMGM manual 'Create Taxonomic Profiling Index tool' (chapter 17.4 for ver. 23).
Examples of combinations related to cell lines and taxonomic names
Prepare bacterial reference genome data.
See 'Downloading the Pathogen Reference Database' in the CMGM manual (Chapter 17.3 for ver. 23).
Run 'Download Pathogen Reference Database' tool included in CMGM.
Download 'All bacteria' data.
To create indexes from reference databases, use 'Create Taxonomic Profiling Index tool'.
Import Illumina read data into an arbitrary folder.
See 'Import high-throughput sequencing data' in the CGW manual (chapter 7.3 for ver. 23).
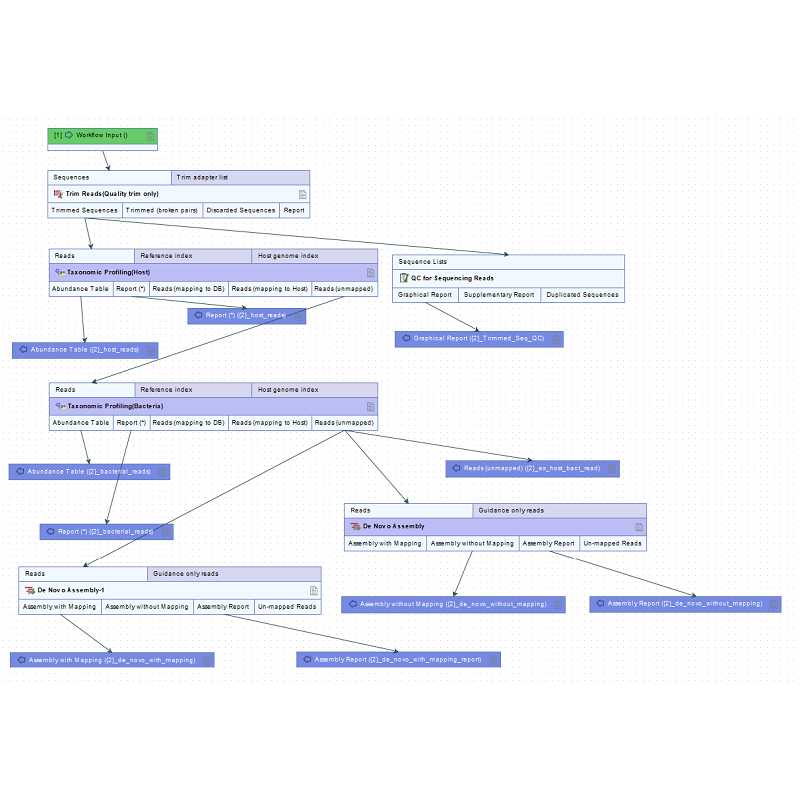
Acquisition of virus-derived contigs from Illumina reads (primary analysis)
Steps 6-9 can be automated using workflow.
The workflow indicated in the image below is an example.
Workflow of primary analysis
Perform quality trimming using the 'Trim reads'.
The Quality limit was set at 0.04.
See 'Trim Reads' in the CGW manual (chapter 26.2 for ver. 23).
Optional
When random amplification using WTA2 kit was performed, you should remove primer sequences derived from the kit.
The primer sequence is comprised from unique and random regions.
The sequence cannot be disclosed due to rights concerns.
For more information, please ask the supplier.
You need to examine the read data and figure out the primer sequence.
The primer sequences can appear in both end of the reads or one of them at various lengths.
See 'Adapter trimming' in the CGW manual (chapter 26.2.2 for ver. 23).
Subtract host derived reads using 'Taxonomic profiling'.
Choose reference index created in step 2.
Unclassified reads which do not map against the host genome are used as input for the next step.
See 'Taxonomic Profiling' in the CMGM manual (chapter 6.2 for ver. 23).
Subtract bacteria derived reads using 'Taxonomic profiling'.
Choose reference index created in step 3.
Reads that do not map against the bacteria genome are used as input for the next step and other subsequent analyses .
Hereafter, the remaining reads is called ex-host-bact-reads (reads excluding host and bacteria)
Assemble ex-host-bact-reads and obtain contigs using 'De novo assembly'.
Select 'Map reads back to contigs' option, and you can obtain a table listing all contigs with consensus length and total read counts of each contig.
Extracting candidate contigs.
Sort the contig list in order of total read counts.
When the library was obtained according to 'Preparation of viral sequencing library for Illumina using NEBNext ultra II', the contig with the highest total read counts is often the target viral genome sequence. Confirm that the consensus length of the contig is close to the size of the viral genome of interest.
Select 10 contigs in order of highest read counts and extract them with 'Extract consensus' tool. These contigs can include one or more viral sequences.
Acquisition of reference nucleotide sequence
Confirm that which of the candidate contigs are viral origin.
Find out viral sequences which are similar with the contigs and are publicly available, using the BLAST at NCBI tool.
See 'BLAST at NCBI' in the CGW manual (Chapter 14.1.1 for ver. 23).
Parameters are modified as below.
Program: blastn, Database: Nucleotide collection, Limit by Entrez query: All organisms, Mask low complexity regions: ‱, Expect: 0.05, Word size: 28, Matrix: Match1/Miasmatch-2, Gap costs: Exist5/Ext2, Max number of hit: 10
Since 10 hits of viral genome sequences similar with a contig are presented by BLAST, you can choose one of them as a reference sequence.
The reference sequence has to be full length, with no recombination region compared to the contig, and having high homology to the contig.
Entering the accession number of the reference sequence into the 'Search for Sequences at NCBI' tool, then download and save it.
Note
The contig obtained by de novo assembly is unreliable at both ends and the endpoint cannot be determined. Therefore, the strategy is to map against the full-length reference sequence to clarify the endpoints. Furthermore, we compare the contig with the mapping consensus using 'align contigs' tool to confirm that there are no discrepancies.
Acquisition of mapping consensus sequence (secondary analysis)
Steps 14-16 can be automated using workflow.
The workflow indicated in the image below is an example.
Workflow of secondary analysis
Map 'ex-host-bact-reads' from step 8 to the reference sequence from step 12 using the 'Map reads to reference' tool.
See 'Map Reads to Reference' tool in the CGW manual (chapter 28.1 for ver.23).
Run the 'Local Realignment' tool to improve on the alignments of the reads in the read mapping.
See 'Local Realignment' tool in the CGW manual (chapter 28.3 for ver.23).
To acquire reliable consensus sequence, run the 'Extract consensus' tool.
Parameters are modified as below.
low coverage threshold: 99 (which adopt as consensus the nucleotides supported by 100 or more reads data)
Low coverage handling: Insert ’N’ ambiguity symbols
See 'Extract consensus' tool in the CGW manual (chapter 28.1 for ver.23).
Sequence comparison of contigs and mapping consensus
Run the 'Align contigs' tool to compare a contig obtained by De novo assemble and a consensus sequence obtained by mapping.
See 'Align contigs' in the CGFM manual (chapter 3 for ver. 23)
Note
If the Align contigs tool finds some mismatches of sequence , open the mapping data obtained in Step 9 and 15 and search for the cause. The cause of mismatch may be the presence of deletions or a large difference due to recombination.
If the mismatch is located near the edge of the genome, the outer parts of the genome might not be compared by the BLAST program. Reducing the BLAST word size or minimum match size may improve analysis.
If necessary, perform the mapping again by changing the reference sequence or manually correct the consensus sequence in the deletion region to resolve the reason for the discrepancy. The final genome sequence is determined based on the mapping consensus.
Protocol references
Yamashita A, Sekizuka T, Kuroda M. VirusTAP: Viral Genome-Targeted Assembly Pipeline. Front Microbiol. 2016 Feb 2;7:32. doi: 10.3389/fmicb.2016.00032.
Qiagen CLC genomics workbench 23.0 manual
Accessed June 13, 2024.
Qiagen CLC Microbial Genomics Module 23.0 manual
Accessed June 13, 2024.
Qiagen CLC Genome Finishing Module 23.0 manual
Accessed June 13, 2024.