Oct 10, 2025
Universal plant and animal eDNA metabarcoding using Nanopore sequencing
- Lucas Esteban Wange1,
- Javier Prado Martinez1,
- Pol Alentorn Moron1,
- Tomàs arquès Bonet2,
- David Juan3,
- Esther Lizano3
- 1Universitat Pompeu Fabra;
- 2iCREA Professor Universitat Pompeu Fabra, Centro Nacional de Análisis Genómico;
- 3Universitat Pompeu Fabra, Centro Nacional de Biotechnología



Protocol Citation: Lucas Esteban Wange, Javier Prado Martinez, Pol Alentorn Moron, Tomàs arquès Bonet, David Juan, Esther Lizano 2025. Universal plant and animal eDNA metabarcoding using Nanopore sequencing. protocols.io https://dx.doi.org/10.17504/protocols.io.dm6gpm2zpgzp/v1
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: October 09, 2025
Last Modified: October 10, 2025
Protocol Integer ID: 229437
Keywords: eDNA, metabarcoding, nanopore, barcoding, real time analysis, biodiversity monitoring, biodiversity assessment, sequencing environmental dna, based biodiversity assessment, tagsteady protocol for multiplexed edna metabarcoding, integrated edna workflow from extraction, animal edna metabarcoding, oxford nanopore library preparation, multiplexed edna metabarcoding, species presence in environmental sample, environmental dna, taxonomic restriction, integrated edna workflow, using nanopore, species presence, simultaneous detection of animal, metazoa, edna, environmental sample, species, oxford nanopore, nanopore, sequencing
Disclaimer
DISCLAIMER – FOR INFORMATIONAL PURPOSES ONLY; USE AT YOUR OWN RISK
The protocol content here is for informational purposes only and does not constitute legal, medical, clinical, or safety advice, or otherwise; content added to protocols.io is not peer reviewed and may not have undergone a formal approval of any kind. Information presented in this protocol should not substitute for independent professional judgment, advice, diagnosis, or treatment. Any action you take or refrain from taking using or relying upon the information presented here is strictly at your own risk. You agree that neither the Company nor any of the authors, contributors, administrators, or anyone else associated with protocols.io, can be held responsible for your use of the information contained in or linked to this protocol or any of our Sites/Apps and Services.
Abstract
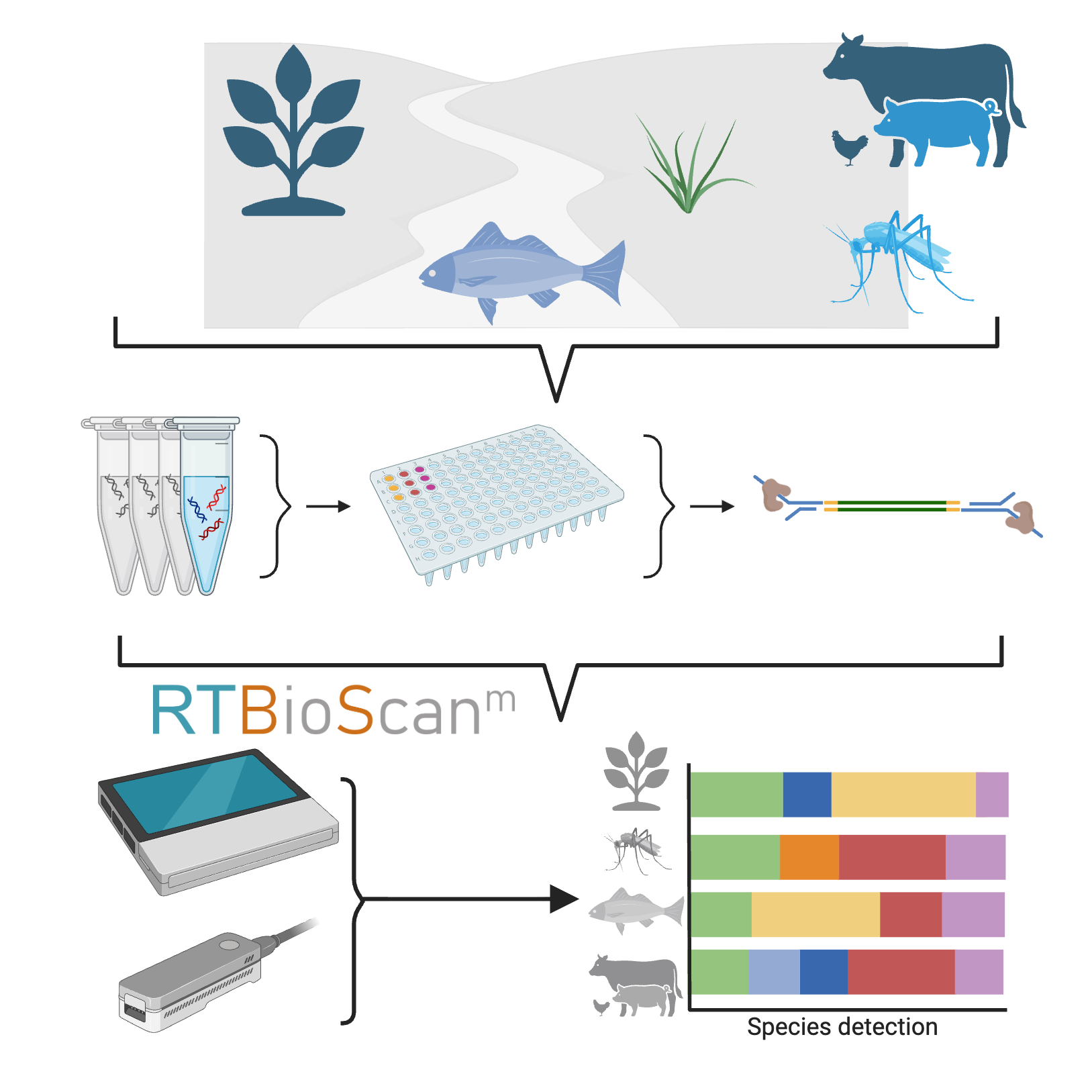
Environmental DNA (eDNA) is a powerful, non-invasive tool for biodiversity monitoring, but traditional approaches face challenges including limited sensitivity, taxonomic restrictions, lengthy processing times, and reliance on specialized lab equipment and computational resources. To address these limitations, we present an integrated eDNA workflow from extraction to real-time analysis. Our approach combines the Tagsteady protocol for multiplexed eDNA metabarcoding with Oxford Nanopore library preparation and sequencing, enabling simultaneous detection of animals (Metazoa) and plants (Viridiplantae) using universal markers (COI and ITS2). By integrating metabarcoding and barcoding in a single protocol, this workflow can assess species presence in environmental samples and barcode individual specimens to expand reference databases, providing a streamlined solution for broad, field-based biodiversity assessment.
Image Attribution
Image partly created with Biorender
Guidelines
It is crucial for the quality of the data obtained to work in the cleanest possible manner. Not only is the targeted DNA present in the sample at a very low concentration, but importantly due to our degenerate primer design almost any animal or plant DNA can be a source of contamination. We suggest to physically separate Pre- and Post-PCR areas and wipe surfaces, gloves and equipment regularly with either diluted bleach or nucleic acid removal solutions like RNAseAway.
Before start
Clean all surfaces, pipettes, centrifuges, magnets and your gloves with RNAseAway before starting. The DNA extraction protocol can be performed comfortably with 16 samples at a once, using a 16 sample tube magnet. Timings in the DNA extraction part assume 16 samples and the use of a repeater pipette to add buffers etc. to samples.
Before Starting
1m
Add 1,040 µl Proteinase K Storage Buffer to each Proteinase K (20 mg) tube prior to use. Store at -20ºC after mixing.
1m
Sample Preparation
52m
Add up to 50 mg material to a bead beating tube containing DNA/RNA Shield and mechanically homogenize your sample on a vortex mixer at maximum speed for 3 minutes.
20m
Add 20 µl Proteinase K and mix well. Incubate at RT for 30 min.l
30m
Centrifuge the sample at 10,000 x g for 1 min to pellet the debris and transfer up to 400 µl of lysate into a new tube.
2m
DNA Purification
1h 58m
Add 400 µl Quick-DNA MagBinding Buffer to 400 µl digested sample and mix by pipetting or inverting 10 times.
5m
Add 33 µl of MagBinding Beads to each sample. If using a repeater pipett use 30 µl
2m
Mix by shaking for 10 min at 1100 – 1500 rpm or by inverting regularly
10m
Transfer the sample to the magnetic rack, wait until beads have separated from the solution
3m
Discard the supernatant using a P1000 pipette
5m
Transfer the sample off the magnetic rack, add 500 µl Quick-DNA MagBinding Buffer.
5m
Mix by shaking or inverting regularly for 10 min
10m
Transfer the sample to the magnetic rack, wait until beads have separated from the solution and discard the supernatant using a P1000 pipette
5m
Transfer the sample off the magnetic rack, add 500 µl DNA Pre-Wash Buffer and mix the sample by pipetting.
5m
Transfer the sample to the magnetic rack, wait until beads have separated from the solution and discard the supernatant using a P1000 pipette
5m
Transfer the sample off the magnetic rack, add 900 µl g-DNA Wash Buffer and mix the sample by pipetting.
5m
Transfer the sample to the magnetic rack, wait until beads have separated from the solution and discard the supernatant using a P1000 pipette
5m
Repeat steps 15-16 two more times.
20m
Air dry for 20 minutes.
20m
Add 40 µl of DNA Elution Buffer to each sample.
3m
Mix by shaking at RT for 5 min.
5m
Transfer the sample to the magnetic rack, wait until beads have separated from the solution and transfer the eluted DNA to a new tube.
5m
Multiplex PCR with Qiagen Multiplex PCR Kit
3h 49m
Set up a no template control reaction (NTC) and if used a positive control containing high quality DNA of animals and/or plants
Note
NTC: 10µl 2x Qiagen Multiplex PCR mix, 6.5 µl H2O
Positive Control: 10µl 2x Qiagen Multiplex PCR mix, 6.5 µl diluted control DNA (max. 1ng total)
2m
Add 19.5 µl eDNA to one well per sample (6.5 per technical triplicate)
5m
Add 30µl 2x Qiagen Multiplex PCR mix to each well
1m
Mix sample and PCR mastermix with a multichanel pipette and distribute 16.5 µl into a total of 3 wells per sample.
5m
Add 3.5 µl of forward reverse barcode mix per well. Or 1.75 µl forward mix and 1.75 µl reverse mix when not using pre-mixed primers. See Notes in "Before you start" section
Note
Use each forward/reverse barcode combination just once per experiment. This is crucial to be able to demultiplex the data from different wells. Note down which primer combination was used for each well meticulously
5m
When using a microwell plate, seal very well to avoid cross contamination.
Note
We recommend using thick plastic seals like MicroAmp clear Adhesive Film
1m
Incubate as indicated below
| A | B | C | D | |
| Step | Temperature | Time | Repeat | |
| Initial denaturation | 95 ºC | 15m | ||
| denaturation | 95 ºC | 30s | 35 cycles | |
| annealing | 55 ºC | 1m 30s | ||
| extend | 72 ºC | 1m 30s | ||
| final extension | 72 ºC | 15m | ||
| store | 12 ºC | Inf |
Multiplex PCR cycling conditions
3h 25m
Choose one normalisation strategy here. Concentration based normalisation gives you more information about the PCR amplification success for each sample, while Bead based normalisation is faster, cheaper and more scalable.
Concentration based normalisation using Qubit59 steps
Here you can choose to either measure ech samples concentration and normalise using this information or adjust concentration using normalisation beads. For recepies of bead normalisation buffer and homemade SPRI beads check "Before you start" section
Bead clean up
38m
Equilibrate SPRI bead solution to RT and vortex until the solution appears homogeneous.
1m
Pool 3 replicates of each sample in one tube/well using a multichannel pipette
2m
Add 1.5x volume beads (with 60 µl of sample, add 90 µl of beads).
5m
Mix 10 times with a pipette and incubate for 5 min.
5m
Place the tubes on the magnet and leave it until the supernatant has cleared.
2m
Discard the supernatant.
1m
Wash twice in 100 µl freshly prepared 80% EtOH.
5m
Air-dry beads for 5 min on the magnet.
5m
Remove the samples from the magnet and add 30 µl ddH2O to the beads.
2m
Mix 10 times with a multichannel pipette and incubate for 5 min.
7m
Put back on the magnet until the supernatant has cleared and transfer the DNA to a new tube.
3m
Concentration measurement
30m
measure 1µl of the cleaned up PCR product using a Qubit flourometer and the Qubit HS DNA Kit according to manufacturers instructions.
5m
In a spreadsheet calculate the amount of DNA to add for each sample. We asume here that the amplicons will have a similar length distribution in all samples.
10m
Note
Typically we aim for 300 fmol total input
Based on the amounts calculated pool amplicons equimolarly and add H2O to 30 µl if necessary.
15m
End Repair
39m
For each library make a master mix:
| A | B | |
| Reagent | Amount | |
| Klenow fragment exo- (5 U/µl) | 1.5 µl | |
| T4 PNK (10 U/µl) | 2 µl | |
| dATP (10mM) | 0.5 µl | |
| T4 DNA ligase buffer (10x) | 4 µl | |
| Reaction booster | 2 µl | |
| Total | 10 µl |
3m
Add 10 µl of master mix to each 30 µl amplicon pool, mix by pipetting and spin down.
1m
Incubate as indicated below:
| A | B | |
| Temperature | Time | |
| 37 ºC | 30m | |
| 65 ºC | 5m | |
| 4 ºC | inf |
35m
Adapter Ligation
46m
Note
Here we use the Oxford Nanopore Library Preparation Kit (Ligation Sequencing Kit V14)
Spin down the Ligation Adapter (LA) and Quick T4 Ligase, and place on ice.
1m
Thaw Ligation Buffer (LNB), Elution Buffer (EB) and Short Fragment Buffer (SFB) at RT and place on ice immediately after thawing and mixing.
1m
Add 20 µl ddH2O to the sample.
1m
In a 1.5 ml DNA LoBind tube, mix in the following order:
| A | B | |
| Reagent | Amount | |
| Endrepaired Amplicon pool | 60 µl | |
| Ligation Buffer (LNB) | 25 µl | |
| NEBNext Quick T4 DNA Ligase | 10 µl | |
| Ligation Adapter (LA) | 5 µl | |
| Total | 100 µl |
Thoroughly mix the reaction by gently pipetting and briefly spinning down.
2m
Incubate the reaction for 10 min at RT.
10m
Resuspend the AMPure XP Beads (AXP).
1m
Add 40 µl of resuspended AMPure XP Beads (AXP) to the reaction and mix by flicking the tube.
1m
Incubate for 5 min at RT.
5m
Spin down the sample, place it on the magnet and pipette off the supernatant when it is clear.
2m
Wash the beads twice with 250 µl Short Fragment Buffer (SFB) and discard the supernatant.
5m
Spin down, place the tube back on the magnet and pipette off any residual supernatant.
2m
Remove the tube from the magnetic rack and resuspend the pellet in 15 µl Elution Buffer (EB).
1m
Spin down and incubate for 10 min at RT.
10m
Pellet the beads on the magnet until the eluate is clear.
1m
Remove 15 µl of eluate and perform concentration measurement using Qubit HS DNA kit according to manufacturers instructions.
1m
Calculate molarity using following formula:
average fragment size for a library containing COI and ITS2 is ~500 bp
Molarity in nM= concentration (ng/µl)/660*mean fragment length (bp)*1e6
µl for 50 fmol= (50 fmol*660* mean fragment length (bp) *1e6)/concentration (ng/µl)
1m
Prepare 50 fmol in 12 µl of Elution Buffer (EB).
1m
Priming and Loading the MinION
21m
Thaw the Sequencing Buffer (SB), Library Beads (LIB), Flow Cell Tether (FCT) and Flow Cell Flush (FCF) at RT and place on ice after thawing.
1m
To prepare the flow cell priming mix with BSA, combine the following reagents:
| A | B | |
| Reagent | Amount | |
| Flow Cell Flush (FCF) | 1170 µl | |
| Bovine Serum Albumin (BSA) at 50 mg/ml | 5 µl | |
| Flow Cell Tether (FCT) | 30 µl |
Note
For detailed instructions on how to load a Minion flow cell consult Nanopores documentation
Open the MinION device lid, slide the flow cell under the clip and press down firmly on the flow cell.
1m
Slide the flow cell priming port cover clockwise to open the priming port.
1m
After opening the priming port, check for a small air bubble under the cover. Draw back a small volume to remove any bubbles:
1m
Set a P1000 pipette to 200 µl.
2m
Insert the tip into the priming port.
Turn the wheel until the dial shows 220-230 µl, to draw back 20-30 µl or until you can see a small volume of buffer entering the pipette tip.
Load 800 µl of the priming mix into the flow cell via the priming port and wait for 5 min.
6m
In a new 1.5 ml DNA LoBind tube, prepare the library for loading as follows:
| A | B | |
| Reagent | Amount | |
| Sequencing Buffer (SB) | 37.5 µl | |
| Library Beads (LB) | 25.5 µl | |
| DNA library (50 fmol) | 12 µl |
3m
Complete the flow cell priming:
2m
Gently lift the SpotON simple port cover to make the SpotON sample port accessible.
Load 200 µl of the priming mix into the flow cell priming port, avoiding the introduction of air bubbles.
Mix the prepared library gently by pipetting up and down just prior to loading.
1m
Add 75 µl of the prepared library to the flow cell via the SpotON sample port in a dropwise fashion.
1m
Gently replace the SpotON sample port cover, making sure the bung enters the SpotON port and close the priming port.
1m
Place the light shield onto the flow cell.
1m
Sequencing and starting the BioScanRT pipeline
Note
Here we assume you have already set up and tested the BioScanRT pipeline following the instructions in github and you have the newest version of MinKnow installed (either on the MinIon device (Mk1c) or on the computer used to control the run (Mk1B or Mk1D)
Run the Flow Cell check. This can also be done already before the Priming and Loading section to save time.
Start the MinKnow run with standard parameters for the Library Preparation Kit (Ligation Sequencing Kit V14), disable bascalling as this will be done on the Laptop or Macbook, depending on library size (<200bp) select to include small fragments
Start the BioScanRT pipeline as outlined in the corresponding github repository.
Protocol references
Carøe, C. & Bohmann, K. Tagsteady: A metabarcoding library preparation protocol to avoid false assignment of sequences to samples. Molecular Ecology Resources 20, 1620–163