Jun 16, 2025
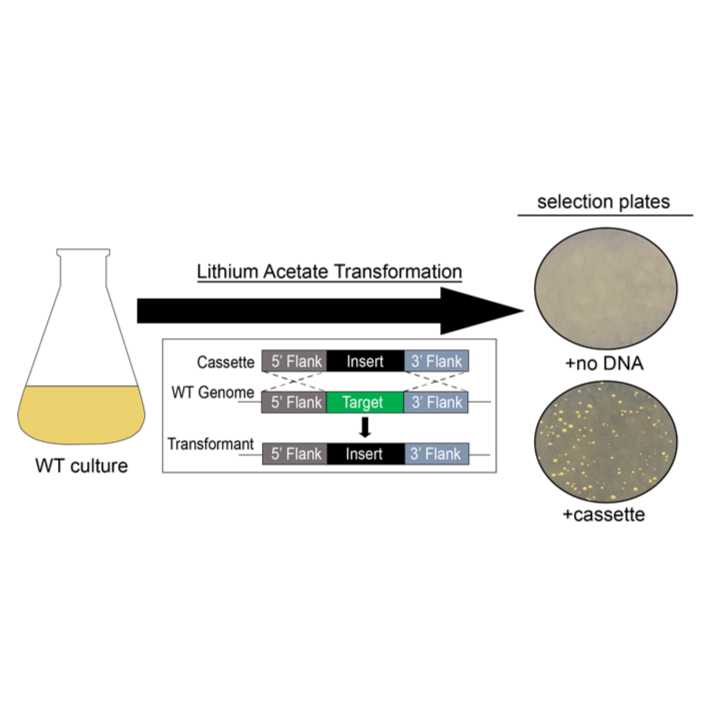
Transformation Protocol for Targeted Homologous Recombination in Auxenochlorella protothecoides
- Marco A Dueñas1,
- Yang-Tsun Lin2,
- Jeffrey Moseley2,
- Sabeeha S. Merchant2
- 1UC Berkeley;
- 2University of California, Berkeley
- Merchant Lab UC Berkeley



External link: https://doi.org/10.1093/plcell/koaf259
Protocol Citation: Marco A Dueñas, Yang-Tsun Lin, Jeffrey Moseley, Sabeeha S. Merchant 2025. Transformation Protocol for Targeted Homologous Recombination in Auxenochlorella protothecoides. protocols.io https://dx.doi.org/10.17504/protocols.io.x54v922mql3e/v1
Manuscript citation:
Craig RJ, Dueñas MA, Camacho DJ, Gallaher SD, Avendaño-Monsalve MC, Lin Y, Blaby-Haas CE, Moseley JL, Merchant SS (2025) Targeted genetic manipulation and yeast-like evolutionary genomics in the green alga Auxenochlorella. The Plant Cell 37(11). doi: 10.1093/plcell/koaf259
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: May 29, 2024
Last Modified: June 16, 2025
Protocol Integer ID: 100870
Keywords: transformation protocol for targeted homologous recombination, auxenochlorella protothecoide, targeted homologous recombination, gene cassettes into the genome, targeted gene cassette, lithium acetate transformation method, auxenochlorella, transformant strain, transformation protocol, genome
Funders Acknowledgements:
Department of Energy (DOE) Office of Science, Biological and Environmental Research
Grant ID: DE-SC0023027
NIH T32 Genetics Dissection of Cells and Organisms Grant
Grant ID: 1T32GM132022-01
Abstract
Detailed instructions for the introduction of targeted gene cassettes into the genome of Auxenochlorella Protothecoides using a lithium acetate transformation method. Following this protocol should yield transformant strains able to be grown on selectable media. This protocol is modified and adapted from Moseley et al. (1) and Dueñas et al. (2).
Materials
Auxenochlorella protothecoides cells
1.5 mL Graduated Flat Cap Tube (Fisherbrand, #02-681-320)
nuclease-free H2O
High-Fidelity‱ restriction enzyme (New England Biolabs)
rCutSmartTM Buffer (New England Biolabs, #B6004S)
Purple Gel Loading Dye, 6X (New England Biolabs, #B7024S)
50 mL Polypropylene Conical Tube (Falcon, #352098)
15 mL Polypropylene Conical Tube (Falcon, #05-539-4)
Polyethylene glycol 4,000 (Alfa Aesar, # A16151)
Tris Hydrochloride (Fisher #BP153-500)
Lithium Acetate dihydrate, 98% (Acros Organics, #6109-17-4)
EDTA (Fisher, #S311-500)
phenol-chloroform-isoamyl alcohol (Fisher #BP17521-400)
Isopropanol (Sigma-Aldrich, #W292907)
Tris-HCl (Sigma-Aldrich, #T5941)
Ethanol (Sigma-Aldrich, #E7023)
Glucose (referred to as Dextrose Anhydrous, Fisher #BP350500)
sterile Milli-Q H2O
Thiamine-HCL (Macron Fine ChemicalsTM, # 0000238814)
ApM1 Media (Refer to )
Safety warnings
phenol-chloroform-isoamyl alcohol 08 (Fisher #BP17521-400) is corrosive. Steps involving this chemical should be done in a hood and with proper PPE and glovewear.
Preparation of DNA for transformation
10m
Before you start, check your designed construct to ensure your gene cassette can be properly digested. Ensure you have restriction sites for digestion at each end of your targeting flanks and no internal restriction sites that will be digested within the cassette.
Digest 100 µg of plasmid construct in 750 µL volume to linearize DNA (usually overnight). Volume of plasmid solution will be dependent on your initial plasmid concentration. It is recommended to perform a maxiprep of your plasmid to ensure adequate plasmid stock. Use the following formula to determine what concentrations to use for your overnight mix:
Plasmid volume to use (Y) = 100000 / (X ng/µL of plasmid)
Overnight digestion mix
Y µL Plasmid solution
75 µL rCutSmartTM Buffer (New England Biolabs, #B6004S)
10 µL of each High-Fidelity‱ restriction enzyme (New England Biolabs)
Z µL of nuclease-free H2O (For adjustment to a final volume of 750 uL)
750 µL Total
Note
If calculated plasmid volume exceeds total, do not add any water and proceed with the maximum volume of plasmid for the 750 µL reaction. Note that this may decrease transformation efficiency.
Run a small aliquot on a gel. Make a mix consisting 3 µL of digestion mix, 2 µL of 6X loading dye ( New England Biolabs, #B7024S), and 10 µL nuclease-free H2O. Ensure complete digestion and proper cassette size.
Note
If digestion appears incomplete, allow restriction digest to proceed for additional time before proceeding,
Example of a linearized DNA for transformation. The top bands represents the gene cassette of interest
Extract digest with 750 µL 25:24:1 phenol-chloroform-isoamyl alcohol 08 (Fisher #BP17521-400) to kill enzyme. Proceed with centrifugation at 10,000 rcf, Room temperature for 00:10:00 .
10m
Transfer aqueous phase (top layer) to a new 1.5 mL tube (Fisherbrand, #02-681-320). Precipitate DNA with 525 µL isopropanol (Sigma-Aldrich, #W292907). Incubate at room temperature for 01:00:00 or overnight at 4 °C .
1h
Air-dry the DNA with cap open for 30-60 minutes in sterile hood. After this step, a small, white pellet should be visible. Dissolve DNA in ~50 µL in nuclease-free H2O or sterile elution buffer (10 millimolar (mM) Tris-HCl (Sigma-Aldrich, #T5941), 7.5 ). DNA should be between 1-2 µg/µL .
Spin down at max speed in microfuge for 00:30:00 . Discard the supernatant and wash the DNA pellet 2 times with500 µL 70 % (v/v) Ethanol (Sigma-Aldrich, #E7023) . Do the wash steps in the sterile hood to avoid contamination. Centrifuge at max speed for 00:05:00 between each wash and remove all traces of Ethanol with a pipette.
35m
Air-dry the DNA with cap open for 30-60 minutes in sterile hood. Dissolve DNA in ~50 µL of sterile elution buffer (10 millimolar (mM) Tris-HCl, 7.5 ). NanoDrop to determine final concentration, which should be between 1-2 µg/µL.
Note
A DNA concentration of 1-2 µg/µL will be adequate for about 3-5 sets transformations. If final concetrations are lower than this amount, transformation can still proceed but ensure to add a higher amount of DNA in step 15.
Making a Transformation Starter Culture
Start a culture of wild-type UTEX 250 cells (or A. pro strain of interest) in 50 mL ApM1 media 26-28 °C in a dark incubator, shaking at 200 rpm for 36:00:00 to 48:00:00 . This can be adjusted as needed but aim for an OD 750 of about 2.0-3.5 (~3 x 107 cells/mL) for the transformation. This can be done from an aliquot as well.
Note
For THIC selection ONLY, the ApM1 start culture should be grown without thiamine added to the media so that the cell culture can deplete residual amounts. To ensure adequate cell concentration and proper thiamine depletion, start the culture with a full loop of cells and allow to grow for 2 days. If this is not done, final transformant cell plates will be overrun and selection will fail.
For information on how to make ApM1 media, plates, glucose stocks, and thiamine stocks, refer to Auxenochlorella ApM1/ApRM1 media recipes and stocks V.2 (dx.doi.org/10.17504/protocols.io.q26g71pq8gwz/v2)
Example of a UTEX 250 transformation starter culture grown in ApM1 2% glucose, 1x thiamine. Pictured in the left flask is a culture just after initial inoculation. On the right is the cells culture after 2 days of growth, with a final OD 750 of 3.5.
3d 12h
Preparing Solutions for Transformation
Prepare the following stock solutions:
Dissolve 90.02 g of Lithium Acetate dihydrate, 98% (Acros Organics, #6109-17-4) to 1 L MQ water to make a 1 Mass Percent Lithium Acetate (LiAC) solution.
Dissolve 1211.4 grams of Tris-HCl (Tris Hydrochloride, Fisher #BP153-500) and 372.24 grams of EDTA (Fisher, #S311-500) in 1L of MQ water to make a 1000x TE Buffer.
Dissolve 500 g of Polyethylene glycol 4,000 (PEG-4000, Alfa Aesar, # A16151) to 500 mL MQ water to make a 50% (w/V) PEG-4000 solution.
Before starting the transformation, make a .1M LiAc/1X TE solution. For a single round of transformation, the following stock is recommended:
LiAc/1xTE Mix (10 mL)
- 1 mL 1 Molarity (M) Lithium Acetate
- 1 mL 1X TE (1m L from 1000x TE stock)
- 8 mL sterile Milli-Q H2O
Note
The lithium acetate solution is critical in increasing permeability of the cell wall for DNA uptake.
Before starting the transformation, make a .40% PEG/.1M LiAc/1x TE solution. For a single round of transformation, the following stock is recommended:
40% PEG/.1M LiAc/1xTE (5 mL)
- 5 mL 1 Molarity (M) LiAc
- 5 mL 1x TE
- 4 mL 50 Mass / % volume PEG-4000
Note
For more than 5 transformations, a 10 mL 40% PEG/.1M LiAc/1xTE solution can be used.
Day 1 of Lithium Acetate Transformation
1h 45m
Transfer the cells to a 50 mL Polypropylene Conical Tube (Falcon, #352098) and centrifuge for at 3750 rpm for 00:10:00 .
10m
Remove supernatant. Add 5 mL of LiAc/1xTE from step 11 to the cell pellet and mix thoroughly while keeping sterile. Repeat centrifugation at 3750 rpm for 00:10:00 .
10m
Remove supernatant and add 500 µL of LiAc/1xTE and mix with a pipette. Transfer this mixture to a 1.7 mL Eppendorf tube (sterile) and allow it to shake for 01:00:00 .
Note
Final amount within the tube will most likely exceed 500 µL due to the volume of the cells and residual solution from the washing step. This should not effect the rest of the protocol and can be used for additional aliquots in step 15.
1h
After 01:00:00 , aliquot 150 µL into eppendorf tubes for each of your plasmid transformations plus a blank. Add DNA cassette to the tube based on the following formula:
DNA to add = 15/(NanoDrop concentration from step 8 /1000).
This should ideally be within a range of 10-20 µL . If higher, use the suggested amount. If lower, use the suggested amount or add 10 µL for simplicity.
In one aliquot, add 15 µL MQ water. This will serve as a transformation blank.
Note
Including the blank will be CRITICAL to ensure the transformation worked properly. Do not skip this step.
1h
Incubate in the shaker for 00:30:00
30m
Add 750 µL of the 40 % (v/v) PEG-400 /.1 Mass Percent LiAc / 1xTE from step 12. Incubate the tubes overnight in a shaker at 26-28 °C .
Note
The PEG solution is critical for DNA permeability, stability, aggregation, and eventual incorporation into the cells.
Overnight incubation of cells in 750 µL of40% PEG-400 / .1 M LiAc / 1xTE solutions.
Day 2 of Lithium Acetate Transformation
8h 0m 30s
For antibiotic selection, add cells to 10 mL ApM1, 2% glucose, 1X thiamine in a 25 mL flask and recover in the dark for ~6-8 hours at24-26 °C , ~120 rpm shaking to allow expression of the antibiotic resistance gene. After this allotted time, transfer cells to 115 mL Polypropylene Conical Tube (Falcon, #05-539-4), and centrifuge at 3750 rpm for00:05:00 and remove the supernatant.
Note
This step is critical in antibiotic selection, skipping it will severely reduce viable colonies or none will appear entirely. For THIC selection, this step can be skipped entirely.
5m
If step 19 was skipped, spin down cells in microfuge at 5000 rpm for 00:00:30 and remove supernatant. For all selections, resuspend cells in 250 µL 1 Mass Percent sorbitol.
Note
The sorbitol supports cell recovery as an osmotic stabilizer after membrane stress and aids in the adherence of cells to the surface of the plates.
30s
Split each transformation between 2 selective plates, ~180 µL per plate. Spread with sterile 4 mm glass beads (Fisher #11-312B) and dry in hood.
For heterotrophic growth, incubate in the dark at 26-28 °C Visible colonies should begin to appear usually after 5-7 days depending on the selection marker. If colonies do not appear after 14 days, the transformation may have failed. Redo and ensure all steps have been followed.
Note
Your colony numbers will vary depending on the method of selection and where your gene cassette is targeted. Generally, you should expect about 10-50+ colonies from a successful transformation plate. You should see close to no colonies (only a few escapes) appear for the blank selection plates.
Example of transformant colonies after 7 days post lithium acetate transformation using a THIC selection marker. Colonies were selected on agarose platesApM1, 2% glucose media lacking any exogenous thiamine. The bottom right plate is a blank where wild type cells were not treated with DNA.
Example of transformant colonies after 7 days post lithium acetate transformation using a neoR selection marker. Colonies were selected on agar plates consisting ApM1, 2% glucose , 1xT Thiamine media with 100 ug/mL of the antibiotic G418. The leftmost plate is a blank where wild type cells were not treated with DNA.
Single Colony Purification of Transformant Colonies
Using a 24 well plate, add 1mL of sterile ApM1 media with 2% glucose. For antibiotic selection, include 1X thiamine and include the antibiotic for further selection. For THIC selection, do not add thiamine
Using either a sterile toothpick or pipette tip, pick 6-12 colonies and mix into a well. Repeat this for 6-12 colonies per strain. We also recommend using a well for a negative control (WT cells) and a positive control (If applicable) to ensure selection media is working as intended.
Allow well cultures to grow for a period of 3-5 days.
Perform dilutions in either a separate 24 well plate or 1.5 mL Eppendorf tubes. Fill each with either sterile MQ water or ApM1 media. For each well plate culture, you will need a set of 2 dilutions
For the dilution 1, add 10 µL of your initial selection culture into the first 1 mL well/tube to make a 1/100 dilution.
Note
Avid mixing for all dilutions steps is CRITICAL to ensure accurate dilutions. This can be done continuously with a P1000 pipette for well plates or capping and shaking with an eppendorf tube.
Take dilution 1 from the previous step and further dilute to make dilution 2 . We recommend a final dilution ranging from ~1/1000 to ~1/5000. See the following amounts for the approximate final dilution:
1/1000: Add 100 uL from dilution 1 into dilution 2
1/2500: Add 40 uL from dilution 1 into dilution 2
1/5000: Add 20 uL from dilution 1 into dilution 2
Note
A higher dilution will ensure more spaced but less colonies. Less dilution will ensure more colonies but tighter spacing for colony picking. If no colonies appear, decrease the dilution amount and repeat single colony purification.
For each dilution, add 200 uL of dilution 2 onto a selection plate and spread using 4 mm glass beads (Fisher #11-312B). Allow to dry for about 30 minutes to an hour, and then wrap and store in the dark in 26-28C. Wait 4-14 days for colonies to appear.
Note
Colony abundance and time of appearance will be dependent on the dilution used. For a lower dilution, expect colonies to appear around 5-10 days post incubation. For a higher dilution, allow up to 14 days for colonies to appear. If no colonies appear after 21 days, redo the single colony purification at a lower dilution.
Protocol references
1. Moseley et al. Production of lipids and terpenoids in Auxenochlorella protothecoides 12 (2021). US Patent US12037630B2, filed November 5th, 2021, granted July 16th, 2024.
2. M.A. Dueñas, R.J. Craig, S.D. Gallaher, J.L. Moseley, & S.S. Merchant,Leaky ribosomal scanning enables tunable translation of bicistronic ORFs in green algae, Proc. Natl. Acad. Sci. U.S.A. 122 (9) e2417695122, https://doi.org/10.1073/pnas.2417695122 (2025).
Acknowledgements
This work was supported by the Department of Energy (DOE) Office of Science, Biological and Environmental Research program under award no. DE-SC0023027 (to J.L.M. and S.S.M.). M.A.D. was supported, in part, by the NIH T32 Genetics Dissection of Cells and Organisms Grant (1T32GM132022-01) and the Newton Graduate Fellowship in Synthetic Biology (QB3-Berkeley).