Jul 31, 2024
Transfections for gene delivery and genome editing in S. rosetta (VERSION 4)
- 1University of California, San Francisco
- Protist Research to Optimize Tools in Genetics (PROT-G)



Protocol Citation: David Booth 2024. Transfections for gene delivery and genome editing in S. rosetta (VERSION 4). protocols.io https://dx.doi.org/10.17504/protocols.io.j8nlk86o5l5r/v1
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: July 31, 2024
Last Modified: July 31, 2024
Protocol Integer ID: 104401
Keywords: cas9 genome editing, execution of crispr, genome editing, crispr, transfections for gene delivery, gene delivery, purified cas9 ribonucleoprotein, guide rna, genome, rna, dna oligonucleotide, dna oligonucleotides as template, cas9 ribonucleoprotein from streptomyces pyogene, cas9 cleavage site, spcas9 rnp, dna, transfection, mutation, gene
Abstract
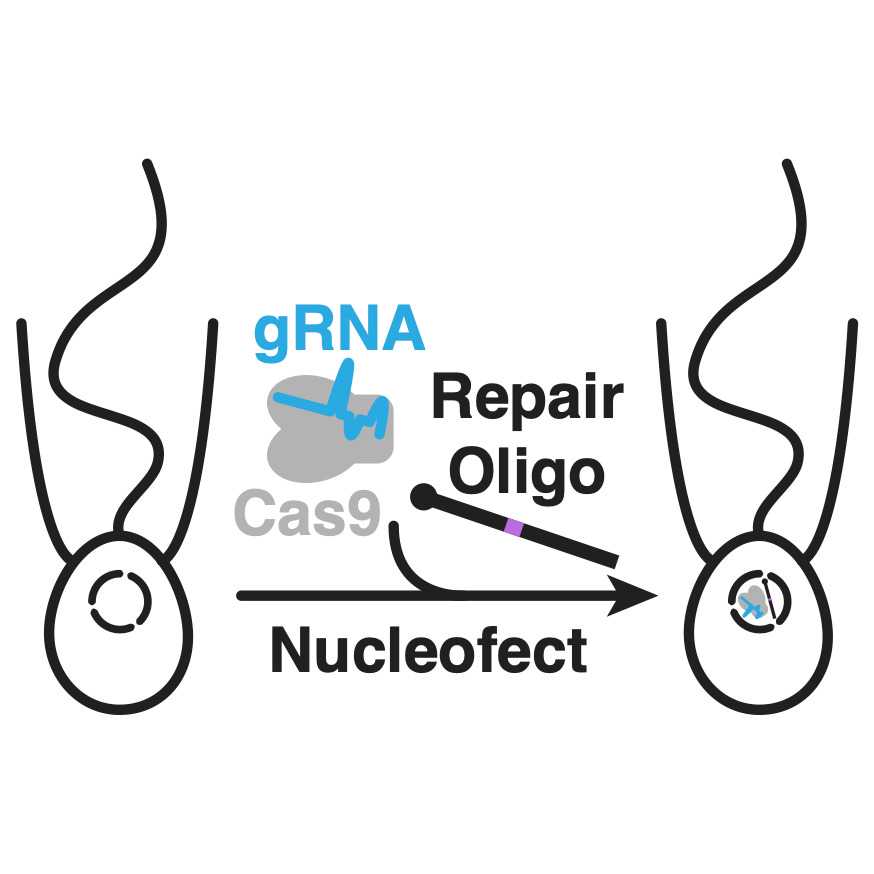
This protocol details the preparation and execution of CRISPR/Cas9 genome editing in S. rosetta. The protocol builds on a method to transfect macromolecules into S. rosetta for delivering a purified Cas9 ribonucleoprotein from Streptomyces pyogenes (SpCas9 RNP) into S. rosetta. Upon cleaving the S. rosetta genome at locations specified by the guide RNA (gRNA) of the SpCas9 RNP, S. rosetta can use DNA oligonucleotides as templates to repair the double-stranded break. Those repair templates can encode foreign sequences and mutations for editing the S. rosetta genome, so long as DNA oligonucleotides have >30 bases of sequence that is homologous to both sides of the Cas9 cleavage site.
Guidelines
Perform cell culturing and transfection procedure inside of a biosafety cabinet to maintain sterility.
Protocol materials
Papain from papaya latexMerck MilliporeSigma (Sigma-Aldrich)Catalog #P3125-100MG
Before start
Please consult the attached file of media recipes for artificial seawater, high nutrient media, and low nutrient media. 20240731_MediaTable.xlsx20.4KB
20240731_MediaTable.xlsx20.4KB
Culture Cells
Seed a large culture of S. rosetta.
Two days prior to transfection, inoculate 80 mL of media (15% Red Algae + 2% Peptone-Yeast-Glycerol) with a culture of S. rosetta feeding on E. pacifica (ATCC PRA-390) to a final concentration of S. rosetta of 1.5*10^4 Mass Percent . Also add 400 µL of 10 Mass Percent of E. pacifica to the flask to provide ample food for growth.
Note
Prepare the E. pacifica solution by resuspending a frozen, 10 mg pellet of E. pacifica in 1 ml of artificial seawater.
Grow the culture for 40:00:00 in a 300 cm2 flask at 22 °C .
1d 16h
Prepare cargo for transgene expression from plasmids
Prepare expression plasmids for transfection
Note
Plasmids can be prepared in advance and stored at -20 °C
Use standard DNA plasmid preparation protocols to purify plasmids from E. coli that lack adenine and cytidine methyl transferases (dam-/dcm-).
If needed, concentrate DNA plasmids to a final concentration of 5 µg/µL using a standard ethanol precipitation.
Note
If you have purified your plasmid with magnetic beads, be sure to thoroughly remove the magnetic beads by centrifuging them at maximum speed for 5 min. Only take the clarified supernatant.
Note
DNA precipitation with ethanol and a salt solution
- Add 3 Mass Percent Potassium Acetate 5.4 to purified plasmid to achieve a final concentration of at least300 millimolar (mM) Potassium Acetate .
- Measure the volume of DNA and potassium acetate and then multiply this volume by 2.5x to calculate the volume of 100% ethanol to add to the DNA/Potassium Acetate mixture.
- After adding the ethanol, thoroughly homogenize the solution. At this point, you may see a white precipitate of DNA.
- Centrifuge the solution at 21000 x g, 4°C, 00:30:00 .
- Remove the supernatant and wash the pellet with 200 µL of 70 % volume Ethanol, ice-cold.
- Centrifuge the pellet at 21000 x g, 4°C, 00:05:00 .
- Remove the supernatant.
- Resuspend the pellet in a minimal volume 10 millimolar (mM) HEPES-KOH 7.5
- Dilute the sample ten-fold in water and read the concentration with a spectrophotometer. 1 µL = 0.05 µg/µL of dsDNA
- Adjust the concentration of the plasmid achieve a final concentration of 5 µg/µL .
Prepare a mixture of carrier molecules to add to the purified plasmid.
Note
The mixture can be prepared ahead of time and stored at -20 °C . Note that the final mixture is very viscous and should be thoroughly homogenized by pipetting up and down before usage.
For one transfection, add 2 µL of 20 µg/µL pUC19 plasmid that has been resuspended in 10 millimolar (mM) Tris-HCl 8.0
For one transfection, add 1 µL of 250 millimolar (mM) Adenosine Triphosphate-KOH 7.5 .
For one transfection, add 1 µL of 100 Mass Percent Heparin, Sodium Salt .
Thoroughly mix the viscous solution by pipetting up and down.
Combine expression plasmids and the carrier mixture for each transfection.
Note
This step can be performed ahead of time, but it is usually done the same day as the transfection.
For one transfection, place 4 µL of the carrier mixture ( ) in the bottom of a 1.5 ml conical tube.
Add 1 µL of of 5 µg/µL Expression Plasmid ( ) to the carrier mixture and slowly pipette up and down to thoroughly mix the solution. This solution is called the "Plasmid Delivery Mix."
Note
The expression of different transgenes may require higher or lower concentrations of plasmid. Performing a titration of plasmid for your particular assay may be necessary. Typically, a range of 1 to 10 µg of plasmid is recommended for one transfection.
Prepare cargo for genome editing with Cas9 RNP
Prepare a guide RNA (gRNA) that binds to SpCas9 and targets DNA by annealing CRISPR RNA (crRNA) with the trans-activating CRISPR RNA (tracrRNA) .
Resuspend crRNA in duplex buffer (30 millimolar (mM) HEPES-KOH 7.5 ; 100 millimolar (mM) Potassium Acetate ) to a final concentration of 200 micromolar (µM) .
Resuspend tracrRNA in duplex buffer to a final concentration of 200 micromolar (µM) .
Mix equal volumes of crRNA ( ) and tracrRNA ( ) to have a final gRNA concentration of 100 micromolar (µM) (gRNA is the annealed complex of crRNA and tracrRNA).
Incubate the gRNA solution at 95 °C in an aluminum block for 00:05:00 .
Place the aluminum block at Room temperature to slowly cool the gRNA to 25 °C .
Store the gRNA at -20 °C .
Prepare DNA oligonucleotides that serve as repair templates after SpCas9 cleavage.
Dissolve oligonucleotides to a final concentration of 250 micromolar (µM) in 10 mM HEPES-KOH, pH 7.5.
Incubate the dissolved oligonucleotides at 55 °C for 01:00:00 .
Store oligonucleotides at -20 °C .
Before starting nucleofections, ensure that the oligonucleotides are fully dissolved by incubating them at 55 °C for 01:00:00 , which concurs while the SpCas9/gRNA complex assembles.
Assemble SpCas9 with the gRNA to form the SpCas9 RNP.
For one transfection, place 2 µL of 20 micromolar (µM) SpCas9 in the bottom of a 0.2 ml PCR tube.
Add 2 µL of 100 micromolar (µM) gRNA ( ) by slowly pipetting up and down with SpCas9 to gently mix the gRNA together. This solution is called the "SpCas9 ribonucleoprotein (RNP)."
Incubate the SpCas9 RNP at Room temperature for 01:00:00 (roughly the time to complete the preparation of S. rosetta for priming, see below).
Prepare transfection Reagents
Prepare SF Buffer (Lonza) for transfections.
Add all of buffer B (smaller volume that may also be called supplement 1) to buffer A (larger volume).
0 µL Store on ice until ready for use. The combined buffer can also be stored at 4°C for up to 3 months.
Note
The combined buffer can be stored at 4°C for up to 3 months.
Note
Because the Lonza kits are quite expensive, we recommend aliquoting large volumes of the SF components (900 µl aliquots for buffer A and 200 µl aliquots for buffer B) to prevent SF buffer from quickly spoiling after buffers A and B have been combined.
Prepare the priming buffer.
Dilute papain to a final concentration of100 micromolar (µM) in dilution buffer (50 millimolar (mM) HEPES-KOH 7.5 ,200 millimolar (mM) Sodium Chloride , 20 % volume Glycerol and 10 millimolar (mM) Cysteine ) from a stock solution of 1 millimolar (mM) Papain (the recommended Papain is already at this concentration), and incubate at Room temperature just before priming cells for transfection.
Papain from papaya latexMerck MilliporeSigma (Sigma-Aldrich)Catalog #P3125-100MG
Note
The dilution buffer should be sterile filtered through a 0.22 µm filter. This buffer may also be prepared ahead of time and stored in a -80°C freezer until just before its use.
Prepare the remaining components of the priming buffer (40 millimolar (mM) HEPES-KOH 7.5 ,34 millimolar (mM) Lithium Citrate , 15 Mass / % volume PEG 8000 and 50 millimolar (mM) Cysteine ). DO NOT combine the papain and priming buffer until just before adding the priming buffer to cells.
Note
The priming buffer without papain should be sterile filtered through a 0.22 µm filter, which can also be made ahead of time and stored at -80°C until it is used. Just be sure that the priming before is warmed to room temperature prior to use.
Wash Cells
Prepare S. rosetta for transfection by washing away feeder bacteria.
Homogenize 80 mL culture of S. rosetta feeding on E. pacifica ( ) by vigorously shaking the flask and then splitting the culture into 40 mL aliquots in 50 ml conical tubes. Vigorously shake the tubes for 30 sec to further break up bacterial clumps and loosely associated S. rosetta cells
Centrifuge the cells at 2400 x g, 4°C, 00:03:00 in a swinging bucket rotor.
Use a serological pipette to gently remove all but 2 ml of the supernatant from the cell pellet. With a fine tip transfer pipette, gently remove the remaining liquid near the pellet. Note that the supernatant may probably be cloudy with E. pacifica bacteria.
Resuspended the two cell pellets in a total volume of 25 mL Cell Wash Buffer , combine into one conical tube, and homogenize the cells by vigorously shaking the tube for 30 sec.
Note
Cell Wash Buffer:
420 mM NaCl
50 mM MgCl2
30 mM Na2SO4
10 mM KCl
titrate to pH 8.0 with ∼2.4 mM NaHCO3
sterile filtered through 0.22 µm filter
For a second time, centrifuge the resuspended cells at 2400 x g, 4°C, 00:03:00 in a swinging bucket rotor.
Remove the supernatant as before ( ).
Resuspend the cell pellet in 100 µL of Cell Wash Buffer . This resuspension is called the "washed cells."
Prepare 100 µL aliquots of 5*10^7 Mass Percent .
Dilute 2 µL of "washed cells" ( ) into 196 µL Artificial Seawater .
Fix the diluted cells with 2 µL of 37.5 Mass / % volume Formaldehyde and homogenize by vortexing.
Pipette the fixed cells into a fixed chamber slide and determine the cell concentration.
Note
Remember that concentration of diluted and fixed cells is a 100-fold dilution from the "washed cells." Be sure to factor that dilution into your concentration.
Note
Cells can be counted on a hemacytometer (Neubauer with brightlines) or with an automated cell counter. We recommend a Luna-FL automated cell counter.
Equipment
LUNA-FL
NAME
Dual Fluorescence Cell Counter
TYPE
Logos Biosystems
BRAND
L20001
SKU
LINK
After determining the cell concentration, dilute the "washed cells" with Cell Wash Buffer to final concentration of 5*10^7 Mass Percent and split into 100 µL aliquots.
Note
One aliquot provides enough cells for 12 nucleofections.
Prime Cells
Prime cells for nucleofection by degrading the glycocalyx that surrounds S. rosetta.
Spin the 100 µL aliquots of washed cells ( ) at 800 x g and 22 °C for 00:02:00 .
Gently remove the supernatant from the cell pellet with a gel-loading pipette tip.
Combine the priming buffer components ( ) to make a final priming buffer (40 millimolar (mM) HEPES-KOH 7.5 ,34 millimolar (mM) Lithium Citrate , 15 Mass / % volume PEG 8000 , 50 millimolar (mM) Cysteine , and 1.5 micromolar (µM) Papain )
Resuspend each cell pellet in 100 µL of priming buffer.
Incubate cells for 00:45:00 at Room temperature .
45m
Add 10 µL of 50 Mass Percent Bovine Serum Albumin to each aliquot of primed cells for quenching proteolysis from the priming buffer.
Centrifuge cells at 1200 x g, 22°C, 00:04:00 .
Gently remove the supernatant from the cell pellet with a gel-loading pipette tip.
Resuspended each cell pell in 25 µL of SF Buffer ( ). This suspension of cells is called the "primed cells."
Store the "primed cells" on ice while preparing nucleofection reactions.
Transfect Cells
Deliver cargo via nucleofection.
For one transfection, add 16 µL of ice-cold SF Buffer to the Plasmid Delivery Mix ( ) or the SpCas9 RNP ( ).
Note
For reactions that use two different gRNAs, assemble each SpCas9 RNP separately then combine each SpCas9 RNP at this step. After the SpCas9 RNPs have been combined, add 16 µL of ice-cold SF Buffer
For genome editing, add 2 µL of the repair oligonucleotide template to the SpCas9 RNP and SF Buffer ( ).
Add 2 µL of "primed cells" (from ) to the tube with delivery cargo and SF Buffer. This solution, which is called the "nucleofection mix."
Transfer the entire nucleofection mix into one well of a 96-well nucleofection plate.
Note
At this point, prepare for the recovery step, by transferring the recovery buffer into a convenient vessel and setting the pipette to 100 µL .
Pulse the nucleofection plate with either the CU 154 (harsher) or the CT 151 (milder) pulse.
Equipment
4D-Nucleofector Core Unit
NAME
Control system for performing nucleofection
TYPE
Lonza
BRAND
AAF-1002B
SKU
LINK
Equipment
96-well Shuttle Device
NAME
Add-on for Nucelofector 4d device to perform plate-based nucleofections
TYPE
Lonza
BRAND
AAM-1001S
SKU
https://bioscience.lonza.com/lonza_bs/US/en/Transfection/p/000000000000191639/96-well-Shuttle-Device
LINK
Rest and Recover Cells
Allow membranes to reseal by resting cells in recovery buffer before growing cells again in media.
Immediately after transfection, add 100 µL of ice-cold recovery buffer (10 millimolar (mM) HEPES-KOH 7.5 ,900 millimolar (mM) Sorbitol , and 8 Mass / % volume PEG 8000 ) to each transfection and gently mixed by firmly tapping the side of the plate.
Note
The recovery buffer should be made ahead of time, sterile filtered through a 0.22 µm membrane, and stored at 4 °C .
Allow cells to rest in recovery buffer for 00:05:00 .
Gently mix the well in the nucleofection plate by pipetting up and down before transferring the entire volume in nucleofection well (the nucleofection mix plus the recovery buffer) into 2 mL 10% Red Algae Media in one well of a 6- well cell culture plate.
Note
Cells may also be transferred into 1 mL 10% Red Algae Media in one well of a 12- well cell culture plate.
Incubate at 27 °C for 00:30:00
Add E. pacifica food and grow transfected cells.
Add 10 µL of 10 Mass Percent of E. pacifica to the wells in the 6-well plate. For a 12-well plate, only add 5 µl.
Note
Prepare the E. pacifica solution by resuspending a frozen, 10 mg pellet of E. pacifica in 1 ml of artificial seawater.
Incubate the cell culture plate at 27 °C for downstream experiments.