May 10, 2023
- Christina Ernst1,
- Julien Duc1,
- Didier Trono1
- 1EPFL - EPF Lausanne



External link: https://doi.org/10.1093/nar/gkad466
Protocol Citation: Christina Ernst, Julien Duc, Didier Trono 2023. TLC-CLIP . protocols.io https://dx.doi.org/10.17504/protocols.io.rm7vzywr4lx1/v1
Manuscript citation:
Ernst C, Duc J, Trono D (2023) Efficient and sensitive profiling of RNA–protein interactions using TLC-CLIP. Nucleic Acids Research 51(13). doi: 10.1093/nar/gkad466
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: August 22, 2022
Last Modified: May 10, 2023
Protocol Integer ID: 68998
Keywords: RNA-Protein interactions, RNA library preparation, Crosslinking and Immunoprecipitation, CLIP, RNA-binding proteins, RBPs, ligation of cdna molecule, profiling of rna, rna molecule, rna, binding protein, improved library preparation strategy for crosslinking, cdna molecule, endogenous rna, improved library preparation strategy, protein interactions in vivo, efficiency of subsequent adapter ligation, subsequent adapter ligation, protein, protein interaction, library preparation strategy, based library preparation strategy, phase cdna
Funders Acknowledgements:
Swiss National Science Foundation
Grant ID: PZ00P3_202048
Human Frontier Science Program
Grant ID: LT000147/2019
Abstract
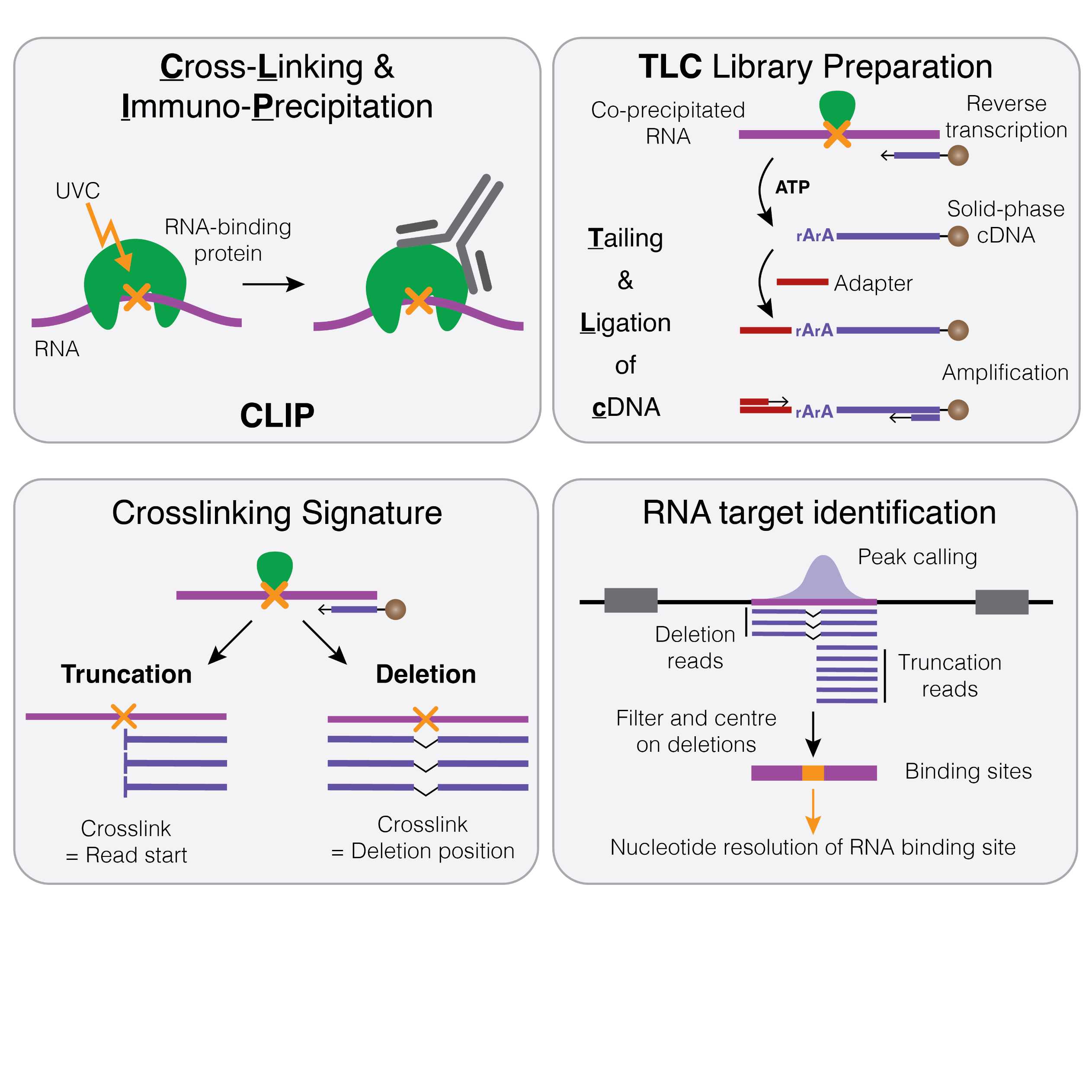
RNA-binding proteins are instrumental for post-transcriptional gene regulation, controlling all aspects throughout the lifecycle of RNA molecules. However, transcriptome-wide methods to profile RNA-protein interactions in vivo remain technically challenging and require large amounts of starting material. Herein, we present an improved library preparation strategy for crosslinking and immunoprecipitation (CLIP) that is based on tailing and ligation of cDNA molecules (TLC). TLC involves the generation of solid-phase cDNA, followed by ribotailing to increase the efficiency of subsequent adapter ligation. These modifications result in a streamlined, fully bead-based library preparation strategy, which eliminates time-consuming purification procedures and drastically reduces sample loss, allowing the profiling of RNA-protein interactions from as few as 1000 cells.
In the accompanying manuscript, we have applied TLC-CLIP to four endogenous RNA-binding proteins, demonstrating its reproducibility and improved precision due to a higher number of crosslinking-induced deletions that serve as an intrinsic quality metric and increase both specificity and nucleotide-resolution.
Guidelines
While working with RNA during the inital parts of the protocol, keep samples cold (on ice) and in RNase-free environment. Use RNase-free water for reactions and buffers.
Materials
Equipment
Equipment
UVP Crosslinker CL-3000
NAME
Analytic Jena
BRAND
UVPA849-95-0615-02
SKU
LINK
Equipment
Branson Tip Sonicator
NAME
Sonicator
TYPE
Branson
BRAND
LPe 40:0.50:4T
SKU
Equipment
ThermoMixer® C
NAME
Eppendorf
BRAND
Catalog No. 2231000680
SKU
LINK
Equipment
SureLock™ Tandem Midi Gel Tank
NAME
Electrophoresis System
TYPE
Invitrogen
BRAND
STM1001
SKU
LINK
Equipment
Criterion Blotter with Plate Electrodes
NAME
Wet-transfer system
TYPE
Bio-Rad
BRAND
1704070
SKU
LINK
Equipment
Odyssey CLx
NAME
Imaging System
TYPE
LI-COR
BRAND
Odyssey CLx
SKU
LINK
Equipment
Bioanalyzer
NAME
Bioanalyzer
TYPE
Agilent
BRAND
G2991AA
SKU
LINK
Any bioanalyzer will suffice.
SPECIFICATIONS
Equipment
Qubit Fluorometer
NAME
Fluorometer
TYPE
Invitrogen
BRAND
Q33238
SKU
LINK
Buffer and Stock Solutions
1M Tris-HCl pH=7.5Invitrogen - Thermo FisherCatalog #15567-027
5M NaClAmbionCatalog #AM9760G
IgepalMerck MilliporeSigma (Sigma-Aldrich)Catalog #I8896
SDS, 10% SolutionLife TechnologiesCatalog #AM9822
Magnesium Chloride Solution BioUltraMerck MilliporeSigma (Sigma-Aldrich)Catalog #68475-100ML-F
Tween 20Merck MilliporeSigma (Sigma-Aldrich)Catalog #P1379-500ml
PEG400Merck MilliporeSigma (Sigma-Aldrich)Catalog #91893
Lithium chloride (8M)Merck MilliporeSigma (Sigma-Aldrich)Catalog #L7026-100ML
EDTA (0.5 M), pH 8.0Life TechnologiesCatalog #AM9260G
UltraPure™ DNase/RNase-Free Distilled WaterThermo FisherCatalog #10977049
Chemicals
Sodium deoxycholate (SDC)Merck MilliporeSigma (Sigma-Aldrich)Catalog #30970
DTT, 1MThermo FisherCatalog #P2325
MethanolFisher ScientificCatalog #code356T
Lithium dodecyl sulfateMerck MilliporeSigma (Sigma-Aldrich)Catalog #L4632
Commercial Kits
Pierce™ Rapid Gold BCA Protein Assay KitThermo FisherCatalog #A53225
ProNex® Size-Selective Purification SystemPromegaCatalog #NG2002
BioAnalyzer High Sensitivity Chip Agilent TechnologiesCatalog #5067-4626
Qubit® dsDNA HS Assay KitThermo Fisher ScientificCatalog #Q32854
Reagents
Dynabeads™ Protein G for ImmunoprecipitationThermo FisherCatalog #10004D
cOmplete™, Mini, EDTA-free (Protease Inhibitor)RocheCatalog ##11836170001)
Oligo(dT)25 DynabeadsThermo Fisher ScientificCatalog #61005
Phusion HF Buffer PackThermo FisherCatalog #F518L
Phusion High-Fidelity PCR Master Mix with HF Buffer - 500 rxns (50 ul vol)New England BiolabsCatalog #M0531L
Enzymes
RNase I (10 U/µL)Thermo FisherCatalog #EN0602
TURBO™ DNase (2 U/µL)Thermo Fisher ScientificCatalog #AM2238
SUPERaseIN RNase InhibitorThermo Fisher ScientificCatalog #AM2696
T4 Polynucleotide Kinase - 2,500 unitsNew England BiolabsCatalog #M0201L
T4 RNA Ligase 1 (ssRNA Ligase) - 5,000 unitsNew England BiolabsCatalog #M0204L
Proteinase K Solution (20 mg/mL)Thermo Fisher ScientificCatalog #AM2546
SuperScript™ IV Reverse TranscriptaseThermo Fisher ScientificCatalog #18090050
Terminal Deoxynucleotidyl TransferaseTakara Bio Inc.Catalog #2230B
T4 RNA Ligase High ConcentrationNew England BiolabsCatalog #M0437
SDS-PAGE
NuPAGE™ LDS Sample Buffer (4X)Invitrogen - Thermo FisherCatalog #NP0008
2-mercaptoethanolMerck MilliporeSigma (Sigma-Aldrich)Catalog #M6250
NuPAGE™ 4-12% Bis-Tris Midi Protein Gels, 20-well, w/adaptersThermo FisherCatalog #WG1402A
NuPAGE™ MOPS SDS Running Buffer (20X)Invitrogen - Thermo FisherCatalog #NP000102
Nitrocellulose Membrane 0.45 umBio-Rad LaboratoriesCatalog #1620115
NuPAGE™ Transfer Buffer (20X)Thermo FisherCatalog #NP00061
Precision Plus Protein All Blue Prestained Protein StandardsBio-Rad LaboratoriesCatalog #1610373
Thick Blot Filter Paper Precut 9.5 x 15.2 cmBio-Rad LaboratoriesCatalog #1704085
Consumables
1.5 mL LoBind tubes EppendorfCatalog #022431021
BRAND(TM) PCR TUBE STRIPS OF 8 ATTACHED SINGLE CAPS STANDARD PROFILE 0.2 MLMerck MilliporeSigma (Sigma-Aldrich)Catalog #BR781332-120EA
RNaseZap®Thermo ScientificCatalog #AM9780
PCR SealsThermo ScientificCatalog #AB0558
Swann-Morton™ Sterile Disposable Stainless Steel ScalpelsFisher ScientificCatalog #11798343
Oligonucleotides
| Name | Sequence | Scale | Purification | Index | |
| 3' Adapter | |||||
| TLC-L3 | /5Phos/AGATCGGAAGAGCACACGTCTGAAAAAAAAAAAAAAAAAAAAAAAAA/3IR800CWN/ | 250nm | RNASE | ||
| 5' Adapter | |||||
| TLC_L01 | /5Phos/NNNNATCACGNNNNNAGATCGGAAGAGCGTCGTG/3ddC/ | 100nm | STD | CGTGAT | |
| TLC_L02 | /5Phos/NNNNCGATGTNNNNNAGATCGGAAGAGCGTCGTG/3ddC/ | 100nm | STD | ACATCG | |
| TLC_L03 | /5Phos/NNNNTTAGGCNNNNNAGATCGGAAGAGCGTCGTG/3ddC/ | 100nm | STD | GCCTAA | |
| TLC_L04 | /5Phos/NNNNTGACCANNNNNAGATCGGAAGAGCGTCGTG/3ddC/ | 100nm | STD | TGGTCA | |
| TLC_L05 | /5Phos/NNNNACAGTGNNNNNAGATCGGAAGAGCGTCGTG/3ddC/ | 100nm | STD | CACTGT | |
| TLC_L06 | /5Phos/NNNNGCCAATNNNNNAGATCGGAAGAGCGTCGTG/3ddC/ | 100nm | STD | ATTGGC | |
| TLC_L07 | /5Phos/NNNNCAGATCNNNNNAGATCGGAAGAGCGTCGTG/3ddC/ | 100nm | STD | GATCTG | |
| TLC_L08 | /5Phos/NNNNACTTGANNNNNAGATCGGAAGAGCGTCGTG/3ddC/ | 100nm | STD | TCAAGT | |
| TLC_L09 | /5Phos/NNNNGATCAGNNNNNAGATCGGAAGAGCGTCGTG/3ddC/ | 100nm | STD | CTGATC | |
| TLC_L10 | /5Phos/NNNNTAGCTTNNNNNAGATCGGAAGAGCGTCGTG/3ddC/ | 100nm | STD | AAGCTA | |
| TLC_L11 | /5Phos/NNNNATGAGCNNNNNAGATCGGAAGAGCGTCGTG/3ddC/ | 100nm | STD | GCTCAT | |
| TLC_L12 | /5Phos/NNNNCTTGTANNNNNAGATCGGAAGAGCGTCGTG/3ddC/ | 100nm | STD | TACAAG | |
| cDNA Amplification | |||||
| P5short_TLC-CLIP | ACACGACGCTCTTCCGATCT | 100nm | PAGE | ||
| P7short_TLC-CLIP | TGACGTGTGCTCTTCCGATCT | 100nm | PAGE | ||
| PCR amplification | |||||
| P5_Universal_adapter | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT | 1um | PAGE | ||
| P7-1_TLC-CLIP | CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC*T | 250nm | PAGE | ATCACG | |
| P7-2_TLC-CLIP | CAAGCAGAAGACGGCATACGAGATACATCGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC*T | 250nm | PAGE | CGATGTAT | |
| P7-3_TLC-CLIP | CAAGCAGAAGACGGCATACGAGATGCCTAAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC*T | 250nm | PAGE | TTAGGCAT | |
| P7-4_TLC-CLIP | CAAGCAGAAGACGGCATACGAGATTGGTCAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC*T | 250nm | PAGE | TGACCAAT | |
| P7-5_TLC-CLIP | CAAGCAGAAGACGGCATACGAGATCACTGTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC*T | 250nm | PAGE | ACAGTGAT | |
| P7-6_TLC-CLIP | CAAGCAGAAGACGGCATACGAGATATTGGCGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC*T | 250nm | PAGE | GCCAATAT | |
| P7-7_TLC-CLIP | CAAGCAGAAGACGGCATACGAGATGATCTGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC*T | 250nm | PAGE | CAGATCAT | |
| P7-8_TLC-CLIP | CAAGCAGAAGACGGCATACGAGATTCAAGTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC*T | 250nm | PAGE | ACTTGAAT |
Safety warnings
Prolonged exposure to UVC light can cause skin and eye damage. Ensure that the UV-crosslinker functions properly and follow all safety precautions specified in the manufacturer's instructions.
Some of the chemicals and reagents used in this protocol can be hazardous if not handled properly. Always follow the safety precautions outlined in the Safety Data Sheets (SDS) provided by the manufacturer.
Before start
Prepare Buffers
iCLIP Lysis Buffer
| Final Concentration | Stock Solution | For 500 ml | |
| 50 mM Tris-HCl, pH 7.4 | 1 M Tris-HCl, pH 7.5 | 25 ml | |
| 100 mM NaCl | 5 M NaCl | 10 ml | |
| 1% Igepal-CA-630 | 100% Igepal CA-630 | 5 ml | |
| 0.1% SDS | 10% SDS | 5 ml | |
| 0.5% sodium deoxycholate | 2.5 g | ||
| Add H2O | 455 ml |
High Salt Wash Buffer
| Final Concentration | Stock Solution | For 500 ml | |
| 50 mM Tris-HCl, pH 7.4 | 1 M Tris-HCl, pH 7.5 | 25 ml | |
| 1 M NaCl | 5 M NaCl | 100 ml | |
| 1 mM EDTA | 0.5M EDTA, pH 8.0 | 1 ml | |
| 1% Igepal-CA-630 | 100% Igepal CA-630 | 5 ml | |
| 0.1% SDS | 10% SDS | 5 ml | |
| 0.5% sodium deoxycholate | 2.5 g | ||
| Add H2O | 364 ml |
PNK Wash Buffer
| Final Concentration | Stock Solution | For 500 ml | |
| 20 mM Tris-HCl, pH 7.4 | 1 M Tris-HCl, pH 7.5 | 10 ml | |
| 10 mM MgCl2 | 2 M MgCl2 | 2.5 ml | |
| 0.2% Tween-20 | 100% Tween-20 | 1 ml | |
| Add H2O | 486.5 ml |
5X PNK Buffer
| Final Concentration | Stock Solution | For 10 ml | |
| 350 mM Tris-HCl, pH 6.5 | 1 M Tris-HCl, pH 6.5 | 3.5 ml | |
| 50 mM MgCl2 | 2 M MgCl2 | 0.25 ml | |
| 5 mM DTT | 1M DTT | 0.05 ml | |
| Add H2O | 6.2 ml |
Freeze individual use aliquots to avoid freeze-thaw cycles.
4X Ligation Buffer
| Final Concentration | Stock Solution | For 10 ml | |
| 200 mM Tris-HCl, pH 7.8 | 1 M Tris-HCl, pH 7.8 | 2 ml | |
| 40 mM MgCl2 | 2 M MgCl2 | 0.2 ml | |
| 4 mM DTT | 1M DTT | 0.04 ml | |
| Add H2O | 7.76 ml |
Freeze individual use aliquots to avoid freeze-thaw cycles.
Proteinase K Buffer
| Final Concentration | Stock Solution | For 50 ml | |
| 100 mM Tris-HCl, pH 7.4 | 1 M Tris-HCl, pH 7.4 | 5 ml | |
| 50 mM LiCl | 8 M LiCl | 0.3125 ml | |
| 1 mM EDTA | 0.5 M EDTA | 0.1 ml | |
| 0.2% LiDS | 1% LiDS | 10 ml | |
| Add H2O | 34.5875 ml |
Oligo(dT) Binding Buffer
| Final Concentration | Stock Solution | For 50 ml | |
| 20 mM Tris-HCl, pH 7.4 | 1 M Tris-HCl, pH 7.4 | 1 ml | |
| 1 M LiCl | 8 M LiCl | 6.25 ml | |
| 2 mM EDTA | 0.5 M EDTA | 0.2 ml | |
| Add H2O | 42.55 ml |
Oligo(dT) Wash Buffer
| Final Concentration | Stock Solution | For 50 ml | |
| 10 mM Tris-HCl, pH 7.4 | 1 M Tris-HCl, pH 7.4 | 0.5 ml | |
| 150 mM LiCl | 8 M LiCl | 0.9375 ml | |
| 0.1 mM EDTA | 0.5 M EDTA | 0.01 ml | |
| Add H2O | 48.5525 ml |
First-Strand (FS) Buffer (5X)
| Final Concentration | Stock Solution | For 10 ml | |
| 250 mM Tris-HCl, pH 8.3 | 1 M Tris-HCl, pH 8.3 | 2.5 ml | |
| 375 mM KCl | 1 M KCl | 3.75 ml | |
| 15 mM MgCl2 | 1 M MgCl2 | 0.15 ml | |
| Add H2O | 3.6 ml |
Generation of preadenylated TLC-L3 adapter
TLC-L3 oligo was ordered from IDT at 250 nmole scale, carrying a 5' phosphorylation and 3' IRDye® 800CW (NHS Ester) (v3) modification and purified using RNase-free HPLC with a total yield of 21.1 nmoles.
- Set up 50 µl of 100 µM TLC-L3 adapter (5 nmoles) with 25 µl 10X 5' DNA Adenylation Reaction Buffer, 25 µl 1 mM ATP and 50 µl Mth RNA Ligase (1nmol) in a total volume of 200 µl using the5’ DNA Adenylation Kit - 50 rxnsNew England BiolabsCatalog #E2610L .
- Incubate at 65ºC for 2 hours followed by inactivation at 85ºC for 10 minutes (reaction turns cloudy).
- Clean up using the Nucleotide Removal KitQiagenCatalog #28304 by mixing the 200 µl preadenylation reaction with 4.8 ml PNI buffer and distributing over 10 columns of Nucleotide Removal Kit.
- Spin down at 6000 rpm for 30 seconds.
- Wash once in 750 µl PE and spin for 1 minute at 6000 rpm, followed by an empty spin at full speed.
- Transfer to a new collection tube and add 50 µl H2O per column and incubate at RT for 2 minutes
- Spin at 6000 rpm for 1 minute to elute
- Combine eluates at an approximate final concentration of 10 µM and prepare 1 µM working stocks to be stored at -20ºC.
UV crosslinking and generation of cell lysates
45m
UV crosslinking and generation of cell lysates
Grow desired cell line to ~80% confluency in appropriate culture conditions.
Remove media, wash once in ice-cold PBS and drain cells of all fluid.
5m
Transfer the plate onto ice, remove lid of the culture dish, and crosslink at 254 nm with 300 mJ/cm2.
On ice
Equipment
UVP Crosslinker CL-3000
NAME
Analytic Jena
BRAND
UVPA849-95-0615-02
SKU
LINK
5m
Scrape cells into 5 ml PBS, count then aliquot desired number of cells and spin down.
10m
Resuspend cell pellet in iCLIP Lysis Buffer - 1 ml buffer for 1 million cells - scale accordingly.
5m
Place lysates on ice for 5 minutes.
On ice 00:05:00
5m
Sonicate lysates for 10-20 seconds at 0.5sec ON and 0.5sec OFF at 10% amplitude to reduce viscosity.
-80 °C Safe Stopping Point
5m
Quantify cell lysates with Pierce BCA Quantification Assay or other method of choice to determine protein concentration.
Expected result
We aim for lysate concentrations of 0.5 µg/µl for CLIP experiments.
Concentrations will differ between samples and cell types - adjust the amount of iCLIP Lysis Buffer used in step 1.4 to your samples to reach the desired concentration.
10m
Preparation of bead-antibody mixture
45m
Preparation of bead-antibody mixture
We routinely use 1-2 µg of antibody per IP in 25-50 µg of cell lysate.
Note
100 µl of protein-G beads bind 20-30 µg of antibody - calculate total volume of beads necessary based on amount of antibody used for experiment.
Wash appropriate amount of beads twice in 1 ml iCLIP Lysis Buffer, then resuspend in 100 µl per antibody
5m
Add antibody to beads and incubate at room temperature for 30-60 minutes rotating.
Room temperature
00:30:00
Note
Continue with section 3 'RNase and DNase Treatment of Lysates' during this incubation step.
30m
Wash beads twice in 1 ml iCLIP Lysis buffer then resuspend in 10 µl 6X Protease Inhibitor per IP.
Note
Add the appropriate amount of antibody-bead mixture to cell lysate to achieve 1X Protease Inhibitor concentration.
5m
RNase and DNase Treatment of Lysates
30m
RNase and DNase Treatment of Lysates
Perform these steps while beads are coupling to antibodies.
Note
Ensure to use the same concentration and the same volume of lysate for a given RBP after optimisation of RNase concentrations.
When optimising RNase concentrations, starting points can be a final amount of 0.25U, 0.025U and 0.005U of RNaseI (EN0602) for 50.000 293T cells (50 µl of lysate at ~0.5 µg/µl).
Note
We aim for a lysate concentration of ~0.5 µg/µl, more concentrated lysates can lead to higher background signal, but can be necessary for lowly expressed RBPs.
Make serial dilutions to achieve desired RNaseI dilution then add 10 µl of RNaseI and 2 µl of Turbo DNase to cell lysates
5m
Digest RNA for exactly 3 minutes at 37°C shaking at 1100 rpm then immediately transfer to ice and incubate for another 3 minutes.
Note
Keep digestion time consistent between experiments to avoid over-digestion of RNA.
10m
Spin lysates for 10 minutes at 4°C at 16,000g then transfer lysates to a fresh tube.
4 °C 00:10:00
15m
Immunoprecipitation and Washes
2h 30m
Immunoprecipitation and Washes
Keep the timing of IP consistent between experiments as RNaseI remains in samples and has residual activity even at 4°C.
5m
Set up 50 µl of RNase-treated lysate with 10 µl of antibody-bead mixture in 6X Protease Inhibitor and incubate for 2 hours at 4°C.
4 °C 02:00:00
2h
Magnetically attract beads and remove supernatant.
Note
Unbound fraction of IP can be kept to test IP efficiency via Western Blot when optimising conditions.
5m
Wash 2 x in 200 µl High Salt Buffer and keep the second wash for at least 1 minute at 4°C.
10m
Wash 2 x in 200 µl PNK Wash Buffer and keep in PNK Wash Buffer until ready to proceed.
10m
Dephosphorylation and first adapter ligation
45m
Dephosphorylation and first adapter ligation
Prepare PNK reaction and add 20 µl per sample.
| PNK Reaction | x 1 | x # | |
| 5X PNK Buffer | 4 µl | ||
| SuperaseIN | 0.5 µl | ||
| T4 PNK | 0.5 µl | ||
| H2O | 15 µl |
10m
Incubate at 37°C for 20 minutes with interval mixing in ThermoMixer.
1800 rpm, 37°C 15 seconds shaking every 2 minutes
Equipment
Eppendorf Thermomixer C Model 5382
NAME
Thermomixer C
TYPE
Eppendorf
BRAND
5382000023
SKU
20m
Remove PNK reaction and wash once in 200 µl PNK Wash Buffer.
5m
Prepare Ligation mix and add 20 µl per sample.
| Ligation Mix | x 1 | x # | |
| 4X Ligation Buffer | 5 µl | ||
| T4 RNA Ligase | 1 µl | ||
| 1 µM L3 Adapter | 1 µl | ||
| SuperaseIN | 0.5 µl | ||
| H2O | 8.5 µl | ||
| PEG400 | 4 µl |
Prepare Ligation Reaction without PEG, mix then add PEG400 and mix by pipetting 10X with P1000.
10m
Incubate at 16°C overnight or at 25°C for 75 minutes with interval mixing in ThermoMixer.
1800 rpm, 16°C 15 seconds shaking every 2 minutes Overnight
Purification of RNA-protein complexes
4h 30m
Purification of RNA-protein complexes
Purification via SDS-PAGE can be omitted to enable a 2-day workflow (see "Omission of PAGE purification"). Please refer to the critical discussion of this step in Ernst et al. (2023) before choosing this workflow.
Remove ligation reaction and wash twice in 200 µl High Salt Buffer and twice in 200 µl PNK Wash Buffer.
Note
When omitting PAGE purification, see step case "noPAGE" for how to proceed.
20m
Resuspend in 20 µl 1X LDS sample buffer containing 5% ß-mercapto-ethanol.
5m
Denature at 70°C for 1 minute then proceed to PAGE purification.
5m
Resolve RNA-protein complexes on NuPAGE 4-12% Bis-Tris Gel at 180V for 60 minutes.
1h 15m
Transfer onto nitrocellulose in 1X NuPAGE transfer buffer with 10% methanol at 30V for 2 hours at RT.
2h 15m
Scan nitrocellulose membrane on Licor infrared scanner with 169 µm resolution.
Expected result
Representative image for hnRNPA1 at three different RNaseI concentrations.
The image shows a representative example of the membrane scan for hnRNPA1 (indicated by a purple triangle), testing three different concentrations for RNaseI (0.25, 0.025 and 0.005U). The image is shown in greyscale (left) and pseudo colouring with higher contrast (right) to show different signal intensities. Purple rectangles indicate the region processed for library preparation.
15m
Place nitrocellulose membrane on filter paper soaked in PBS to cut out region of interest and place nitrocellulose pieces in 1.5 ml LoBind Tubes.
Note
Region of interest usually corresponds to ~20-60 kDa above the molecular weight of the RBP of interest due to the ligation of TLC-L3 adapter (~15.9 kDa) and additional weight depending on the length of associated RNA molecule (70 nt of RNA are on average 20kDa).
15m
Omission of PAGE purification53 steps
- When omitting PAGE purification, magnetic beads bound to RNA-protein complexes in step 6.2 can be directly resuspended in 100 µl Proteinase K buffer containing 100 µg of Proteinase K and incubated at 50°C at 800 rpm for 45 minutes (see Step 7.1).
- Attract beads and transfer supernatant to fresh PCR tubes containing oligo(dT) beads (see Step 7.4).
RNA Purification
1h 30m
RNA Purification
Upon capture on oligo(dT) beads (Step 7.5), resuspend beads throughout all steps by vortexing unless otherwise stated. Depending on the reaction, oligo(dT) beads can be sticky and mixing by pipetting can cause unnecessary loss of material through retention in pipette tips.
Add 200 µl of Proteinase K buffer containing 100 µg of Proteinase K to LoBind tube containing nitrocellulose pieces and incubate at 50°C at 800 rpm for 45 minutes.
800 rpm, 50°C, 00:45:00
Note
1 µl of Proteinase K reaction can be dot blotted on nitrocellulose to visualise RNA release.
1h
Meanwhile, prepare 10 µl of oligo(dT) beads per sample and wash once in 1 ml oligo(dT) Binding Buffer.
10m
Resuspend oligo(dT) beads in 50 µl Binding Buffer per sample and distribute in fresh PCR tubes.
5m
Transfer Proteinase K reaction to oligo(dT) beads and incubate at RT for 10 minutes rotating.
15m
Wash twice in 125 µl oligo(dT) Wash Buffer and once in 20 µl 1X First-Strand (FS) Buffer by vortexing.
15m
Reverse Transcription
50m
Reverse Transcription and RNA elution
Prepare RT reaction and add 10 µl to beads - vortex to mix.
| Reverse Transcription | x1 | x # | |
| 5X FS Buffer | 2 µl | ||
| 10mM dNTPs | 0.5 µl | ||
| 0.1M DTT | 0.1 µl | ||
| SuperaseIN | 0.3 µl | ||
| Superscript IV | 0.1 µl | ||
| H2O | 7 µl |
10m
Incubate at 50°C for 15 minutes with interval mixing then heat up to 96°C on thermomixer.
Note
Seal tubes with PCR plates seal to avoid lids from opening at higher temperatures on heatblock.
25m
When 96°C is reached, vortex for 30 seconds on heatblock, spin down, and immediately place on magnet on ice.
5m
Remove supernatant and wash beads once in 60 µl oligo(dT) Wash buffer and once in 20 µl 1X T4 RNA Ligase buffer - vortex to mix.
Note
Supernatant can be kept to visualise the elution of RNA-TLC-L3 hybrid through dot blotting on nitrocellulose.
Expected result
Example of dot blots at different steps during the protocol to visualise the amount of adapter-ligated RNA.
10m
Second adapter ligation
50m
Second adapter ligation
Add 5 µl of 5' adapter mix to beads and vortex to mix.
| 5' Adapter Mix | x1 | x # | |
| 10X T4 RNA Ligase Buffer | 2 µl | ||
| 10 µM L## oligo | 2 µl | ||
| DMSO | 1 µl |
Note
Ensure balanced nucleotide composition of in-read barcodes that are used at this step, by using at least four different adapters that, when multiplexed, have a balanced 'per base sequence content' for the barcode sequence.
5m
Incubate at 75°C for 2 minutes then immediately place on ice.
5m
Add 4 µl of Ligation mix at the top of the tube, spin down and vortex to mix.
| Ligation mix | x1 | x # | |
| 0.1 M ATP | 0.5 µl | ||
| TdT (14U/µl) | 0.5 µl | ||
| T4 RNA Ligase High Conc. (30U/µl) | 0.5 µl | ||
| H20 | 2.5 µl |
Note
Beads can be sticky, avoid touching with pipette tip when adding ligation mix!
5m
Add 10 µl PEG8000, spin down and vortex, then resuspend beads by pipetting up and down 10X at slow speed.
10m
Incubate at 37°C for 20 minutes, then cool down to 20°C.
37 °C 00:20:00
20m
Add 1 µl of T4 RNA Ligase (High Concentration) and pipette to mix before incubating overnight at 20°C.
Overnight 20 °C
5m
cDNA pre-amplification
1h
cDNA pre-amplification
Add 100 µl of oligo(dT) Wash buffer to ligation reaction and place on magnet.
Note
Beads don't resuspend properly at this point - apply magnetic field from different sides of the tube until beads move swiftly from one side to the other in order to aid resuspension.
5m
Discard supernatant and wash once more with 100 µl oligo(dT) supernatant and once in 20 µl 1X Phusion HF Buffer - vortex to mix.
10m
Prepare cDNA pre-amplification mix and resuspend beads in 25 µl - vortex to mix.
| cDNA amplification | x1 | x # | |
| 2X Phusion HF PCR Mastermix | 12.5 µl | ||
| 10 µM P7&P5 short | 1.25 µl | ||
| H2O | 11.25 µl |
5m
Amplify with the following programme:
1. 98°C - 30 seconds
2. 98°C - 10 seconds
3. 65°C - 30 seconds
4. 72°C - 30 seconds
Go to Step #2 6 times
5. 72°C - 3 minutes
6. 16°C - HOLD
Note
When preparing libraries after omission of PAGE purification a total of 6 pre-amplification cycles are sufficient.
20m
Wash 1-2 µl of oligo(dT) beads per sample in oligo(dT) Binding buffer and resuspend in 5 µl oligo(dT) binding buffer per sample.
5m
Add 5 µl of oligo(dT) beads to PCR reaction and rotate at RT for 5 minutes.
10m
Place on magnet and transfer amplified cDNA in supernatant to a fresh tube.
Note
Pre-amplified cDNA can be stored at 4°C for short-term or -20°C for longer term.
5m
cDNA size selection
50m
cDNA size selection
Perform ProNex Size Selection using 2.8X ProNex beads - add 84 µl of ProNEX beads and mix by pipetting up and down 10 times.
Note
The exact ratio of ProNEX beads might have to be optimised for different batches of beads.
5m
Incubate for 10 minutes at RT then place on magnet for 5 minutes.
15m
Discard supernatant and wash twice in 200 µl Wash Buffer with beads remaining on the magnet.
5m
Remove Wash buffer and air-dry the beads on magnet until pellet starts to show cracks.
10m
Resuspend in 23 µl H2O and incubate for 5 minutes at RT.
10m
Place on magnet and transfer 20 µl of supernatant to a new tube.
Expected result
Pre-amplified cDNA can be run on High Sensitivity Bioanalyser Chip as anadditional quality control step, but will most likely only show signal for strong RBPs.
5m
PCR amplification
1h 45m
PCR amplification
Estimate the necessary number of PCR cycles for ideal library amplification by running a test qPCR on 1 µl of pre-amplified cDNA and subtract 3-4 cycles from the obtained Ct value for the final library amplification. Overamplification of TLC-CLIP libraries should be avoided to minimise the number of PCR duplicates and increase the final yield of usable reads.
Prepare qPCR Mastermix and add 9 µl to 1 µl of cDNA.
| Test qPCR | x 1 | x # | |
| 2X PowerUP SYBR Green Mastermix | 5 µl | ||
| 10 µM P5 + P7 primers | 0.5 µl | ||
| H2O | 3.5 µl |
Determine optimal cycle number by running the following programme:
1. 98°C - 30 seconds
20 cycles of:
2. 98°C - 10 seconds
3. 68°C - 30 seconds
4. 72°C - 30 seconds
Note
Calculate required PCR cycles for amplification by removing 3-4 cycles from the determined Ct value.
Expected result
Amplification curves for duplicate libraries from different starting material.
1h
Mix 10 µl of cDNA with 30 µl of PCR Mastermix and amplify with the following programme:
| PCR | x 1 | x # | |
| 2X Phusion HF Mastermix | 20 µl | ||
| 10 µM P5 + P7 primers | 1 µl | ||
| H2O | 9 µl |
1. 98°C - 30 seconds
# of cycles:
2. 98°C - 10 seconds
3. 68°C - 30 seconds
4. 72°C - 30 seconds
5. 72°C - 3 minutes
6. 16°C - HOLD
Note
Add different i7 indexes at this point to allow greater multiplexing.
Note
10 µl of cDNA is used for PCR amplification to allow repetition in case of substantial over-amplification or unexpected size profile.
45m
Size-selection of libraries
50m
Size-selection of libraries
Perform ProNex Size Selection using 1.8X ProNex beads - add 72 µl of ProNEX beads and mix by pipetting up and down 10 times.
5m
Incubate for 10 minutes at RT then place on magnet for 5 minutes.
15m
Discard supernatant and wash twice in 200 µl Wash Buffer while on magnet.
5m
Air-dry the beads on magnet until pellet starts to show cracks
10m
Resuspend in 17.5 µl H2O and incubate for 5 minutes at RT.
10m
Place on magnet and transfer 15 µl of supernatant to a new tube.
5m
Quality Control and Quantification
1h
Quality Control and Quantification
Prepare 1:3 or 1:5 dilution for QC and quantification.
Run 1 µl on Agilent High Sensitivity DNA Chip.
Expected result
1h
Quantify libraries using Qubit High Sensitivity dsDNA kit.
20m
Multiplexing and sequencing
Multiplexing and sequencing
Multiplex samples in the final pool at the desired ratios and sequence on Illumina NextSeq500 using the High Output Kit for 75 cycles with the addition of 5% PhiX.
Demultiplexing and Trimming
Demultiplexing and Trimming of Reads using Flexbar
This is done in a two-step approach for the following reason:
The easiest way to trim 3' adapter contamination is to specifcy the entire adapter sequence (see Flexbar_2 below) with 'N' being used for the i7 index positions so that this step does not have to be run individually for different i7 indeces.
However, if this is specified together with --umi-tags, any bases on the 3' end that might correspond to the i7 index will be added to the read header and appended to the UMI which interferes with umi-tools as some UMIs will have more characters than others.
As such it is easiest to handle the demultiplexing based on In-read barcodes as well as the UMIs in the first step, and then trim remaining homopolymers at the 5' end as well as adapter contamination at the 3' end in a second step.
Demultiplexing based on In-read barcodes
Flexbar.v3.4 was used.
This command demultiplexes based on in-read barcodes and moves UMIs in the read header.
-r sample.fastq.gz # Fastq file demultiplexed based on i7 index but not trimmed
-b TLC_barcodes.fasta # Fasta file specifying in-read barcodes and UMIs
--barcode-unassigned # Generates file containing all unassigned barcodes
--barcode-trim-end LTAIL # Defines barcode position within the read
--barcode-error-rate 0 # Determines number of mismatches and indels allowed in barcode
--umi-tags # Wildcard character 'N' specified within barcode fasta file will be appended to read name separated by underscode
-n 4 # Number of threads
-t path/sample_demult # Prefix of output files
-z GZ # Output files are compressed using gzip
Command
Flexbar_1
Example of TLC_barcodes.fasta file
Command
Fasta file containing in-read barcodes for demultiplexing with Flexbar.
TLC_barcodes.fasta
Trimming of adapter and homopolymers
flexbar.v3.4 was used
This command trims remaining homopolymers at the 5' end of reads as well as adapter contamination at the 3' end.
-r sample_demult.fastq.gz # Demultiplexed fastq files from previous trimming step
--adapter-seq 'AGATCGGAAGAGCACACGTCTGAACTCCAGTCACNNNNNNATCTCGTATGCCGTCTTCTGCTTG'
# Seqeunce of the Illumina Multiplexing Index Read Sequencing Primer with 'N' designating the position of different i7 indices.
--adapter-trim-end RIGHT # Defines which end of the read to trim the adapter sequence off
--adapter-error-rate 0.1 # Determines how many mismatches and indels are allowed for the adapter sequence to be removed
--adapter-min-overlap 1 # Minimum required overlap for adapter to be removed
--min-read-length 18 # Discards reads shorter than 18 nucleotides after trimming
-n 2 # Number of threads
--htrim-left T # Trims poly(T) from the left side of the read
--htrim-max-length 2 # Defines maximum length of poly(T) stretch to be trimmed as 2
--htrim-min-length 1 # Defines minimum length of poly(T) stretch as 1
-t path/sample_tr1-2 # Prefix of output files
-z GZ # Output files are compressed using gzip
Command
Flexbar_2
Remove space from read header before mapping to keep UMI
Command
new command name
Mapping and Deduplication
Mapping is performed with STAR, keeping only uniquely mapping reads and removing the penalty for opening deletions or insertions.
STAR mapping
STAR v.2.7.3a was used
--runThreadN 8 # Number of threads
-genomeDir /genome_index/STAR/ # Path to genome directory
--readFilesIn sample_tr1-2.fastq.gz # Path to fastq file
--readFilesCommand zcat # Uncompession Command for compressed fastq
--outSAMtype BAM SortedByCoordinate # Output sorted by coordinate
--limitBAMsortRAM 20000000000 # Maximum available RAM for sorting BAM
--outSAMattributes All # Include all attributes
--outSAMunmapped Within # Output unmapped reads within the main SAM file
--runRNGseed 42 # random number generator seed
--outTmpDir {localdir} # path to temporary directory
--outFilterMultimapNmax 1 # maximum number of multiple alignments allowed for a read
--outSJfilterReads Unique # Only uniquely mapping reads are considered for splice junction output
--alignEndsType Extend5pOfRead1 # Fully extend only the 5' end of the read, local alignment for 3' end
--scoreDelOpen 0 # Remove deletion open penalty
--scoreInsOpen 0 # Remove insertion open penalty
--outFileNamePrefix path/sample # Prefix for output files
Command
STAR_mapping
Deduplication with umi-tools
UMI-tools v.1.0.1 was used in dedup mode:
-I mapping/star/sample_tr1-2.bam # mapped bam file to read
-L logs/umi_tools/umi_tools_sample.log # file with logging information
-S mapping/star_dd/sample_dd.bam # output file
--extract-umi-method read_id # Barcodes are contained at the end of the read header
--method unique # Reads group share the exact same UMI
--spliced-is-unique # Two reads that start in the same position on the same strand and having the same UMI are considered unique if one is spliced and the other is not.
Command
umitools_dedup