Oct 25, 2023
Targeted detection of SNCA CNVs in SOX10+ nuclei from oligodendrocytes containing alpha-synuclein inclusions isolated from human post-mortem brain
- Caoimhe Morley1,
- Diego-Perez Rodriguez2,
- Monica Emili Garcia-Segura2,
- Ester Kalef-Ezra2,3,
- Christos Proukakis2,3
- 1University College London;
- 2Department of Clinical and Movement Neurosciences, Queen Square Institute of Neurology, University College London, London, UK;
- 3Aligning Science Across Parkinson’s (ASAP) Collaborative Research Network, Chevy Chase, MD, 20815



Protocol Citation: Caoimhe Morley, Diego-Perez Rodriguez, Monica Emili Garcia-Segura, Ester Kalef-Ezra, Christos Proukakis 2023. Targeted detection of SNCA CNVs in SOX10+ nuclei from oligodendrocytes containing alpha-synuclein inclusions isolated from human post-mortem brain. protocols.io https://dx.doi.org/10.17504/protocols.io.5jyl8p6r7g2w/v1
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working.
Created: October 09, 2023
Last Modified: May 31, 2024
Protocol Integer ID: 89876
Keywords: ASAPCRN, targeted detection of snca cnv, vulnerable to genomic mosaicism, somatic cnvs in specific cell population, genomic mosaic, snca gene of patient, synucleinopathy, synuclein inclusion, snca gene, current genomic technology, genomic mosaicism, human genome, level genomic mosaic, nuclear oligodendrocyte marker, dna fluorescence in situ hybridisation, nuclei from oligodendrocyte, fish with immunofluorescence, genome, dna fluorescence, specific protein marker expression, oligodendrocyte, dna, parkinson, multiple system atrophy, alternative cytogenetic method, specific cell population, complex neurodevelopmental, gene, such as parkinson, immunofluorescence
Funders Acknowledgements:
Multiple System Atrophy Trust
Grant ID: 2022/93020 (572015)
The Michael J. Fox Foundation for Parkinson’s Research (MJFF) and the Aligning Science Across Parkinson’s (ASAP)
Grant ID: 000430
Disclaimer
This protocol was adapted from the following:
Garcia-Segura, M.E., Perez-Rodriguez, D. and Proukakis, C. (2022) ‘Combined fluorescence in situ hybridization (FISH) and immunofluorescence for the targeted detection of somatic copy number variants in Synucleinopathies’, Neuromethods, pp. 229–243.
Ester Kalef-Ezra, Diego Perez-Rodriguez, Christos Proukakis. Manual isolation of nuclei from human brain using CellRaft device and single nucleus Whole Genome Amplification. Protocols.io (https://protocols.io/view/manual-isolation-of-nuclei-from-human-brain-using-cx4mxqu6).
Abstract
There has been a growing recognition of the complexity of the human genome, and the role somatic variation plays in disease. The brain is particularly vulnerable to genomic mosaicism, likely arising during complex neurodevelopmental and ageing processes. However, current genomic technologies often lack the sensitivity to detect low-level genomic mosaics that could contribute to disease. An alternative cytogenetic method is DNA fluorescence in situ hybridisation (FISH), which allows for a targeted analysis of rare, disease-relevant copy number variants (CNVs). FISH can be subsequently combined with immunofluorescence to characterize somatic CNVs in specific cell populations based on specific protein marker expression. This protocol describes a method combining FISH with immunofluorescence, which we name immuno-FISH, for the detection of CNVs in the SNCA gene of patients with synucleinopathies, such as Parkinson’s disease (PD) and Multiple System Atrophy (MSA). This method is performed on nuclei isolated from frozen, human post-mortem brain tissue, which addresses potential sectioning artefacts and reduces protease digestion for epitope preservation. Our protocol is optimised to detect SOX10, a nuclear oligodendrocyte marker, and alpha-synuclein inclusions, which are frequently retained at the perinucleus in MSA (the so-called Papp-Lantos inclusions). This protocol also describes its use in affected PD and MSA brain regions such as the putamen, substantia nigra (SN) and cerebellum.
Attachments

862-2224.pdf
611KB
Guidelines
Intended purposes:
This protocol has been optimised for use on single-nuclei isolated from flash-frozen, human post-mortem brain tissue. It can be adapted to different SureFISH Agilent probes and antibodies for detecting nuclear markers.
Figure 1. Overview of nuclei isolation and immuno-FISH protocol (created using BioRender).
Materials
Equipment
- Tissue culture hood for human sample handling
- PCR Laminar Flow Cabinet
- Refrigerated centrifuge for 1.5mL tubes capable of reaching 13,000xg
- Oven capable of maintaining 37°C for FISH hybridisation
- Water bath capable of reaching 72°C
- P1000, P200, P20, P2 Pipettes with filtered tips
- Fume hood
- Pair of forceps and scissors
- Haemocytometer
- Dounce tissue grinder set 2mL (Kimble via Sigma Aldrich – D8938)
Table 1. Specifications of reagents used for nuclei isolation method from human post-mortem brain tissue.
| A | B | C | D | |
| Item | Supplier | Catalogue Ref. | Preparation prior use | |
| UltraPure DNase/RNase-Free Distilled Water | Thermo Fisher | 10977049 | Aliquot and keep at RT | |
| PBS (Phosphate Buffered Saline) 10X Solution (pH 7.4) | Thermo Fisher | 15815418 | Make 1x with dH2O and store at 4°C | |
| 50x cOmplete Protease Inhibitor Cocktail EDTA-free | Roche via Sigma Aldrich | 4693159001 | Use 1 tablet in 1 ml dH2O and store at -20°C | |
| Triton-X100 | Sigma Aldrich | T9287 | Prepare 10% aliquot and store at RT | |
| ODGM (Optiprep Density Gradient Medium) | Sigma Aldrich | D1556 | Aliquot and keep at 4°C | |
| Dithiothreitol (DTT) | Prepare 1 mM and keep aliquots at -20°C | |||
| Sucrose | Prepare 1 M and keep at -20°C |
Table 2: Specifications of the consumables used for immuno-FISH protocol.
| A | B | C | |
| Item | Supplier | Catalogue Ref. | |
| EasyDip™ slide staining system | Simport | M905-12DGY | |
| SuperFrost Ultra Plus™ GOLD Adhesion Slides | Epredia™ | 11976299 | |
| Glass coverslips 22mm x 22mm | VWR | 631-0124 | |
| Glass coverslips 22mm x 50mm | VWR | 631-0137 | |
| FixoGum Rubber Cement | Marabu | 29010017000 | |
| Nail Varnish | |||
| 1.5mL Polypropylene DNA LoBind Microcentrifuge Tubes | Eppendorf™ | 0030108418 | |
| 0.2mL PCR Tubes | Eppendorf™ | 951010006 |
Table 3. Specifications of reagents used for immuno-FISH protocol.
| A | B | C | D | |
| Reagent Name | Supplier | Catalogue Ref. | Preparation prior to use | |
| Methanol >99.5% Pure | Thermo Fisher | M/4000/21 | No | |
| Glacial Acetic Acid | Thermo Fisher | BP1185 | No | |
| Magnesium Chloride Hexahydrate, BioXtra, ≥99.0% | Sigma-Aldrich | M2670 | Dissolve 1 M in dH2O and store at RT | |
| Pepsin 1g from porcine gastric mucosa | Sigma | D1000 | Prepare 10% solution and store in aliquots at -20°C | |
| 1M Hydrochloric acid (HCl) | Thermo Fisher | 124210025 | No | |
| UltraPure™ Formamide | Thermo Fisher | 15515026 | No | |
| 20X SSC Buffer, Molecular Grade | Promega | V4261 | 2X solution in dH2O and stored at RT | |
| UltraPure™ DNase/RNase-Free Distilled Water | Thermo Fisher | 10977049 | No | |
| Molecular Grade 100% Ethanol (EtOH) | Thermo Fisher | BP2818 | Prepare solutions of 70%, 90% and 100% EtOH and store one at RT and one at -20°C | |
| SureFISH hybridisation buffer | Agilent | G9400A | No | |
| SureFISH custom-designed probe 50kb SNCA 4q22.1 | Agilent | G110902G-8 | No | |
| SureFISH Wash Buffer 1 | Agilent | G9401A | No | |
| SureFISH Wash Buffer 2 | Agilent | G9402A | No | |
| SureFISH Chr7 CEP probe 767kb P20 GR | Agilent | G110899G-8 | No | |
| Goat Serum | Sigma Aldrich | G9023 | Store in aliquots at -20°C | |
| Triton™ X-100, BioXtra | Merck | T9284 | Prepare 0.2% Solution in 1X PBS stored at RT | |
| PBS Tablets | Life Technologies | 18912014 | Prepare 1X with dH2O stored at RT | |
| DAPI (4’, 6-diamidino-2-phenylindole, Dihydrochloride) | Sigma-Aldrich | D9542 | Prepare 1 mg/mL aliquots stored at -20°C | |
| TrueBlack® Lipofuscin Autofluorescence Quencher | Biotium | 23007 | No | |
| Prolong™ Gold Anti-Fade Mountant | Thermo Fisher | P36930 | No |
Table 4. Specifications of antibodies used in this immuno-FISH protocol.
| A | B | C | D | |
| Antibody | Species | Supplier | Catalogue Ref. | |
| Primary antibodies | ||||
| SOX10 (SP267) | Rabbit | Abcam | Ab227680 | |
| a-Syn (Syn 211) | Mouse | Santa-Cruz | sc-12767 | |
| Secondary antibodies | ||||
| Anti-Rabbit Alexa Fluorophore 647 | Goat | Thermo-Fisher | A21245 | |
| Anti-Mouse Alexa Fluorophore 488 | Goat | Thermo-Fisher | A11001 | |
Table 5. FISH pre-treatment solutions.
| A | B | C | D | |
| Solution Name | Reagents | Volume | Final Concentration | |
| Pepsin solution | 10% Pepsin aliquot | 50 μL | 0.01% | |
| dH2O | 100 mL | |||
| 1M HCl | 1000 μL | 10 mM | ||
| PBS/MgCl2 solution | 1X PBS | 100 mL | ||
| 1M MgCl2 | 100 μL | 1 mM | ||
| Formamide solution | 99.5% Formamide | 70 mL | 70% | |
| 2M SSC | 30 mL | 0.6 M |
Table 6. FISH probe mixture per 22 x 22 mm reaction area / slide.
| A | B | C | |
| Reagent | Volume (μL) | Final % concentration | |
| Custom-designed SureFISH probe 50kb SNCA 4q22.1 - Fluorophore 568 | 1 | 10 | |
| SureFISH Chr7 CEP probe 767kb P20 GR – Fluorophore 488 | 1 | 10 | |
| SureFISH Hybridisation buffer | 7 | 70 | |
| Nuclease-free H2O | 1 | 10 | |
| Total | 10 |
Table 7. Immunofluorescence solutions.
| A | B | C | D | |
| Solution Name | Reagent | Volume (μL) | Final concentration | |
| Blocking Solution | Goat serum | 30 | 10% | |
| 0.2% Triton-X in 1X PBS | 270 | |||
| Primary Antibody solution | Rabbit anti-SOX10 | 3 | 0.5 μg/mL | |
| Mouse anti-Syn 211 | 0.75 | 1 μg/mL | ||
| Goat serum | 3 | 2% | ||
| 0.2% Triton-X in 1X PBS | Adjust to 150 μL | |||
| Secondary Antibody solution | Goat Anti-Rabbit Fluorophore 648 | 0.3 | 2 μg/mL | |
| Goat Anti-Mouse Fluorophore 488 | 0.3 | 2 μg/mL | ||
| Goat serum | 3 | 2% | ||
| 0.2% Triton-X in 1X PBS | Adjust to 150 μL | |||
| TrueBlack solution | 20X TrueBlack Lipofuscin quencher | 10 | 1X | |
| 70% EtOH | 190 |
UltraPure™ DNase/RNase-Free Distilled WaterThermo FisherCatalog #10977049
PBS (Phosphate Buffered Saline) 10X Solution (pH 7.4)Fisher ScientificCatalog #15815418
cOmplete mini EDTA free protease inhibitor cocktailMerck MilliporeSigma (Sigma-Aldrich)Catalog #4693159001
Triton™ X-100Merck MilliporeSigma (Sigma-Aldrich)Catalog #X100-5ML
OptiPrep™ Density Gradient MediumMerck MilliporeSigma (Sigma-Aldrich)Catalog #D1556)
Methanol, Certified AR for AnalysisThermo Fisher ScientificCatalog #M-4000-21
Acetic Acid, Glacial (Aldehyde-Free/Sequencing), Fisher BioReagents™™Thermo Fisher ScientificCatalog #BP1185-500
Magnesium chloride hexahydrateMerck MilliporeSigma (Sigma-Aldrich)Catalog #M2670
Pepsin from porcine gastric mucosaMerck MilliporeSigma (Sigma-Aldrich)Catalog #P7000
Hydrochloric acid, 1N standard solutionThermo Fisher ScientificCatalog #124210025
UltraPure™ FormamideThermo FisherCatalog #15515026
SSC Buffer, 20X, 1LPromegaCatalog #V4261
UltraPure™ DNase/RNase-Free Distilled WaterThermo FisherCatalog #10977049
Ethanol, Absolute, Molecular Biology GradeThermo Fisher ScientificCatalog #BP2818500
FISH Hybridization BufferAgilent TechnologiesCatalog #G9400A
FISH Wash Buffer 1Agilent TechnologiesCatalog #G9401A
FISH Wash Buffer 2Agilent TechnologiesCatalog #G9402A
Goat serumMerck MilliporeSigma (Sigma-Aldrich)Catalog #G9023
Triton™ X-100Catalog #T9284
PBS TabletsThermo FisherCatalog #18912014
4′,6-Diamidino-2-phenylindoleMerck MilliporeSigma (Sigma-Aldrich)Catalog #D9542
TrueBlack® Lipofuscin Autofluorescence QuencherBiotiumCatalog #23007
ProLong™ Gold Antifade MountantThermo FisherCatalog #P36930
Recombinant Anti-SOX10 antibodyAbcamCatalog #ab227680
Anti-α-synuclein Antibody (211)Santa Cruz BiotechnologyCatalog #sc-12767
Goat anti-Rabbit IgG (H L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 647Thermo Fisher ScientificCatalog #A-21245
Goat anti-Mouse IgG (H L) Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 488Thermo Fisher ScientificCatalog #A-11001
Safety warnings
Safety warnings
All tissue cutting and nuclei isolation steps must be performed in a Class II biosafety cabinet. Toxic chemicals such as formamide must be used in a fume hood. Refer to the SDS of each reagent for details on handling guidelines.
Nuclei isolation from human post-mortem brain tissue using iodixanol gradient
Set the centrifuge to 4 °C .
Prepare ice-cold Carnoy’s fixative (3:1 Methanol: Glacial acetic acid) and 1X PBS.
Isolate nuclei manually:
See Table 1 for reagents and steps used for nuclei isolation. Refer to Kalef-Ezra, Perez-Rodriguez and Proukakis (dx.doi.org/10.17504/protocols.io.kxygxzjjov8j/v1) for details of the methods and solutions required for nuclei isolation implemented here.
Tissue guidelines: Use approximately 10-50 mg of brain tissue per nuclear suspension. Nuclei yield will vary between samples due to tissue collection, disease progression, and sub-regional differences between grey and white matter (cellular density and lipid composition among others).
Note
Notes
- For the putamen, 20-50 mg of tissue is recommended. For the cerebellum and substantia nigra, 10-30 mg is recommended due to overall higher cellular density and proportion of lipid content within these regions.
- The granular layer of the cerebellar cortex cannot be fully disassociated by Dounce homogenisation and may cause clumps within the nuclear suspension.
Nuclear yield check and visualization with DAPI (optional)
30m
Resuspend the pellet containing the isolated nuclei in 500 µL of DAPI (1 µL in 1x PBS working concentration).
Leave the tube on a rotator disk for 00:20:00 at 4 °C .
20m
Centrifuge at 800 x g, 4°C, 00:05:00 and remove the supernatant.
5m
Resuspend in 100-200 µL of 1X PBS.
Use a haemocytometer and an epifluorescence microscope to estimate yield and visualise the spread of nuclei. The nuclear suspension should be evenly distributed, appear as single nuclei and free of large debris (see Figure 2 for examples).
Figure 2. Examples of isolated nuclei stained with DAPI illustrating (A) areas of nuclei clumping and (B) evenly distributed, single nuclei.
Centrifuge at 800 x g, 00:05:00 to pellet nuclei and remove the supernatant.
5m
Nuclei fixation and preparation onto slide
1h 15m
Resuspend pellet containing the isolated nuclei in 1mL of pre-chilled Carnoy’s fixative and leave to fix on a rotator disk for 01:00:00 at 4 °C .
1h
Centrifuge 800 x g, 00:05:00 and remove the supernatant.
5m
Resuspend pellet in 100-200 µL of Carnoy’s fixative by pipetting up and down.
Optional: A 70 µm Flowmi cell strainer can be used to filter large clumps and debris.
Using a dropper or a pipette, place nuclear suspension onto an EprediaTM SuperFrost Plus Gold Adhesion slide and leave to evaporate for 20-60 min Room temperature .
Note
- The charge of the SuperFrost Plus Gold Adhesion Slides repels PBS, therefore we do not recommend dropping a nuclear suspension containing PBS as it will require hours to evaporate and forms crystallised salts on the slide.
- We recommend using a Super PAP pen to create a hydrophobic barrier prior to dropping the nuclei to contain the nuclear suspension within a small area on the slide.
Wash slides.
Wash slides for 00:05:00 at Room temperature in an EasyDip slide staining jar containing 1X PBS. (1/2)
5m
Wash slides for 00:05:00 at Room temperature in an EasyDip slide staining jar containing 1X PBS. (2/2)
5m
Check under microscope to assess the spread of nuclei before proceeding with immuno-FISH.
FISH Pre-treatment
1h 25m
Prepare slide staining jars for FISH Pre-treatment according to Table 5 of Materials.
Place the water bath in a fume hood and set it to 72 °C .
Submerge the staining jar containing formamide solution into the water bath.
Set the oven to 37 °C and place the jar with dH2O inside (to which pepsin will be added afterwards), allow at least 00:30:00 for solutions to reach the desired temperature.
30m
Add HCl and pepsin to dH2O jar (according to Table 5 of Materials), then immediately place the slides in the pepsin solution for 00:05:00 in the oven.
5m
Transfer the slides to the PBS/MgCl2 solution and leave for 00:05:00 at Room temperature .
5m
Wash with 1X PBS once for 00:05:00 at Room temperature .
5m
Dehydrate the isolated nuclei in increasing concentrations of EtOH.
Dehydrate the isolated nuclei in 70% EtOH stored at Room temperature for 00:02:00 .
2m
Dehydrate the isolated nuclei in 90% EtOH stored at Room temperature for 00:02:00 .
2m
Dehydrate the isolated nuclei in 100% EtOH stored at Room temperature for 00:02:00 .
2m
Allow the slides to air-dry for 00:10:00 on the bench at Room temperature .
Note
Note: In the meantime, take out the FISH probes and hybridisation buffer from -20 °C to equilibrate to Room temperature , taking care to avoid exposure to direct light.
10m
Incubate the slides in the formamide solution for 00:03:00 at 72 °C .
3m
Dehydrate the nuclei in EtOH (pre-chilled at -20 °C ).
Dehydrate the nuclei in 70% EtOH (pre-chilled at -20 °C ) for 00:02:00 at
Room temperature .
2m
Dehydrate the nuclei in 90% EtOH (pre-chilled at -20 °C ) for 00:02:00 at
Room temperature .
2m
Dehydrate the nuclei in 100% EtOH (pre-chilled at -20 °C ) for 00:02:00 at
Room temperature .
2m
Allow the slides to air-dry for 00:10:00 on the bench at Room temperature .
Note
Notes
- In the meantime, prepare the FISH probe mixture as outlined in Table 6 of Materials.
- This protocol can be performed as a 1-colour or 2-colour FISH probe reaction depending on the number of protein markers being investigated. If two protein markers will be used for immunofluorescence, the reference probe can be excluded, and the volume of the reaction mix adjusted with Nuclease-free H2O.
10m
Denature the FISH probe mixture for 00:05:00 at 72 °C in the water bath.
5m
Add 10 µL of the probe mixture to the slide, evenly distributing small droplets onto the nuclear suspension.
Place a 22mm x 22mm coverslip and seal the edges with rubber cement.
The FISH probes can be left to hybridise to DNA in a humidified box kept in the dark at 37 °C for 48-96 hrs.
FISH Post-hybridisation treatment & immunofluorescence staining
30m
Note
Prepare immunofluorescence solutions according to Table 7 of Materials.
Place the water bath in a fume hood, then set temperature to 72 °C .
Add Wash Buffer 1 at least 00:30:00 in the water bath.
30m
Take out an aliquot of goat serum from -20 °C and leave to thaw at Room temperature .
Peel off the rubber cement manually, soak the slides in 2X SSC for 00:10:00 and then remove the coverslips from the slides.
10m
Wash the slides in FISH Wash Buffer 1 for 00:02:00 at 72 °C in the water bath.
2m
Wash the slides in FISH Wash Buffer 2 for 00:01:00 at Room temperature .
1m
Wash the slides.
Wash the slides 00:10:00 in 1X PBS at Room temperature . (1/3)
10m
Wash the slides 00:10:00 in 1X PBS at Room temperature . (2/3)
10m
Wash the slides 00:10:00 in 1X PBS at Room temperature . (3/3)
10m
Hand-dry sections with tissue to remove PBS excess and create a hydrophobic barrier around the section using a Super PAP pen.
Note
Notes
- Be careful not to damage the nuclei on the slides.
- If the barrier pen was previously used for containing the nuclear suspension, apply more in the same area.
Add 300 µL of the blocking solution and leave the slides in a humidified chamber for 01:00:00 at Room temperature or Overnight at 4 °C .
2h
Remove the blocking solution excess and apply 150 µL of the primary antibody solution.
Leave to incubate 2-4 hrs at Room temperature or Overnight at 4 °C .
1h
Wash the primary antibody solution off.
Wash the primary antibody solution off in 1X PBS for 00:10:00 at Room temperature . (1/3)
10m
Wash the primary antibody solution off in 1X PBS for 00:10:00 at Room temperature . (2/3)
10m
Wash the primary antibody solution off in 1X PBS for 00:10:00 at Room temperature . (3/3)
10m
Add 150 µL of the secondary antibody solution and leave to incubate for 01:00:00 at Room temperature .
1h
Wash the secondary antibody off.
Wash the secondary antibody off in 1X PBS for 00:10:00 at Room temperature . (1/3)
10m
Wash the secondary antibody off in 1X PBS for 00:10:00 at Room temperature . (2/3)
10m
Wash the secondary antibody off in 1X PBS for 00:10:00 at Room temperature . (3/3)
10m
Add 1 µL DAPI (working concentration) to the slides for 00:20:00 .
20m
Wash in 1X PBS for 00:05:00 at Room temperature .
5m
Add 200 µL of TrueBlack solution for 00:01:00 .
1m
Quickly rinse the slides with 70% EtOH and then wash.
Wash the slides in 1X PBS for 00:10:00 at Room temperature . (1/3)
10m
Wash the slides in 1X PBS for 00:10:00 at Room temperature . (2/3)
10m
Wash the slides in 1X PBS for 00:10:00 at Room temperature . (3/3)
10m
Add 10-20 µL of Prolong Gold Anti-Fade solution and mount a 22mm x 22mm coverslip.
Leave the slides to dry in the dark Overnight at Room temperature before sealing the edges of the coverslip with nail varnish. Store them at 4 °C until use.
Note
Notes
- In our experience, nuclear suspension autofluorescence can interfere with FISH signal detection, and so we have incorporated a quenching treatment step.
- For optimal acquisition, suspensions can be imaged within 2 weeks on any 4-colour fluorescence microscope with resolution to detect small FISH signals. We use 16 Z-stacks of 0.5uM to capture focal planes across the nucleus.
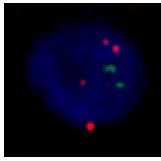
Figure 3. Examples showing (a) Chr 7 CEP and SNCA FISH signals and (b) a SOX10+ nucleus.
10m