Jul 29, 2023
SNARE-seq2 with Nuclei Hashing
Forked from SNARE-seq2
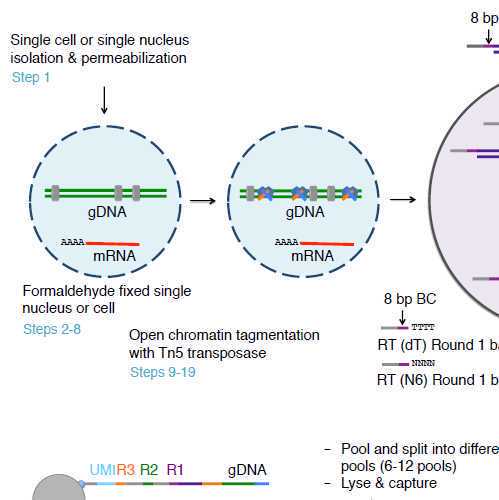
- Nongluk Plongthongkum1,2,
- Dinh H Diep1,
- Song Chen1,
- Blue Lake1,
- Kun Zhang1
- 1University of California, San Diego;
- 2King Mongkut's University of Technology Thonburi
- Human BioMolecular Atlas Program (HuBMAP) Method Development CommunityTech. support email: [email protected]



Protocol Citation: Nongluk Plongthongkum, Dinh H Diep, Song Chen, Blue Lake, Kun Zhang 2023. SNARE-seq2 with Nuclei Hashing . protocols.io https://dx.doi.org/10.17504/protocols.io.eq2lyjn4qlx9/v1
Manuscript citation:
Plongthongkum N, Diep D, Chen S, Lake BB, Zhang K. Scalable dual-omics profiling with single-nucleus chromatin accessibility and mRNA expression sequencing 2 (SNARE-seq2). Nat Protoc. 2021 Oct 14. doi: 10.1038/s41596-021-00507-3. Epub ahead of print. PMID: 34650278.
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: July 29, 2023
Last Modified: November 23, 2023
Protocol Integer ID: 85647
Keywords: seq2 with nuclei hashing, nucleus chromatin accessibility, nucleus rna, chromatin barcode ligation, joint profiling of gene expression, chromatin accessibility, nuclei hashing, rna, transcription, mrna in the cytoplasm, reverse transcription, sequencing library, mrna, mrna expression, captured cdna, nucleus, gene expression, cell barcode ligation, same nucleus, seq2, next rounds of cell barcode ligation, nuclei, nucleus atac, joint profiling, snare, whole cell, complex tissue, cdna, cell, cytoplasm, seq2 with nuclei
Disclaimer
This is a modification of the original published protocol.
Abstract
To study the heterogeneity of complex tissues by joint profiling of gene expression and its regulation, we require an accurate and high-throughput method. Here we described improved high-throughput combinatorial indexing-based single-nucleus chromatin accessibility and mRNA expression sequencing 2 (SNARE-Seq2) co-assay. This protocol involves fixing and permeabilizing the nucleus followed by tagmentation, chromatin barcode ligation, reverse transcription, pooling and splitting for the next rounds of cell barcode ligation into cDNA and accessible chromatin (AC) on the same nucleus. The captured cDNA and AC are co-amplified before splitting and enrichment into single-nucleus RNA and single-nucleus AC sequencing libraries. The protocol can also be applied to both nuclei and whole cells to capture mRNA in the cytoplasm. This improvement allows us to generate hundreds of thousands of data set of each assay and can be scaled up to half a million cells from a single experiment. The entire procedure can be complete in 3.5 d for generating joint single-nucleus RNA and single-nucleus ATAC sequencing libraries.
Attachments

Materials
MATERIALS
In-house Tn5 transposase or alternatively, Tagmentase (Unloaded)DiagenodeCatalog #C01070010-20
Tagmentase Dilution BufferDiagenodeCatalog #C01070010
T4 DNA Ligase Reaction Buffer - 6.0 mlNew England BiolabsCatalog #B0202S
NEBuffer 3.1 - 5.0 mlNew England BiolabsCatalog #B7203S
Hemo KlenTaq - 1,000 rxnsNew England BiolabsCatalog #M0332L
T7 DNA Ligase - 750,000 unitsNew England BiolabsCatalog #M0318L
PMSFMerck MilliporeSigma (Sigma-Aldrich)Catalog #P7626
T4 DNA LigaseNew England BiolabsCatalog #M0202
Tween 20Merck MilliporeSigma (Sigma-Aldrich)Catalog #P9416-50ML
Microseal® ‘B’ Adhesive SealsBio-Rad LaboratoriesCatalog #MSB-1001
DNA LoBind Tube 1.5ml EppendorfCatalog #022431021
RNAse InhibitorEnzymaticsCatalog #Y9240L
Tango BufferThermo Fisher ScientificCatalog #BY5
twin.tec PCR Plate 96 LoBind semi-shirted clear 25 pcs.EppendorfCatalog #30129504
Dynabeads MyOne Streptavidin C1Invitrogen - Thermo FisherCatalog #65001
Ficoll PM-400 20% in H2OMerck MilliporeSigma (Sigma-Aldrich)Catalog #F5415-50ML
Qubit assay tubesThermo Fisher ScientificCatalog #Q32856
Qubit dsDNA HS Assay KitThermo Fisher ScientificCatalog #Q32854
cOmplete™ Protease Inhibitor CocktailMerck MilliporeSigma (Sigma-Aldrich)Catalog #11697498001
Low DNA Mass LadderThermo FisherCatalog #10068013
UltraPure™ DNase/RNase-Free Distilled WaterThermo FisherCatalog #10977015
Pierce™ 16% Formaldehyde (w/v), Methanol-freeThermo FisherCatalog #28906
SUPERase• In™ RNase Inhibitor (20 U/μL)Thermo FisherCatalog #AM2696
GlycoBlue™ Coprecipitant (15 mg/mL)Thermo FisherCatalog #AM9515
ATP Solution (100 mM)Thermo FisherCatalog #R0441
Bovine Albumin Fraction V (7.5% solution)Gibco - Thermo Fisher ScientificCatalog #15260037
PBSGibco - Thermo Fisher ScientificCatalog # 10010023
Polyethylene Glycol 6000 (PEG 6000)Merck MilliporeSigma (Sigma-Aldrich)Catalog #81255-1KG
Tris hydrochloride (1M) pH 8.0Thermo Fisher ScientificCatalog #15568025
Magnesium chloride solution for molecular biology (1.00 M)Merck MilliporeSigma (Sigma-Aldrich)Catalog #M1028
Potassium chloride (2M)Thermo Fisher ScientificCatalog #AM9640G
5 M Sodium chloride (NaCl)Merck MilliporeSigma (Sigma-Aldrich)Catalog #S5150-1L
NN-Dimethylformamide (DMF)Merck MilliporeSigma (Sigma-Aldrich)Catalog #227056
Advantage UltraPure dNTP combination kit (100 mM each dNTP) Takara Bio Inc.Catalog #639132
DL-DithiothreitolMerck MilliporeSigma (Sigma-Aldrich)Catalog #10708984001
Sodium dodecyl sulfate solutionThermo Fisher ScientificCatalog #AM9822
Maxima H Minus Reverse Transcriptase (200 U/uL)Thermo Fisher ScientificCatalog #EP0753
Bovine Serum Albumin (20 mg/mL) Molecular Biology GradeNew England BiolabsCatalog #B9000S
EDTA (0.5 M) pH 8.0Merck MilliporeSigma (Sigma-Aldrich)Catalog #20158
Triton X-100 Merck MilliporeSigma (Sigma-Aldrich)Catalog #X100-100ML
Proteinase K solution (20mg/mL) RNA gradeThermo Fisher ScientificCatalog #25530049
SSC Buffer (20X)Merck MilliporeSigma (Sigma-Aldrich)Catalog #S6639
KAPA HiFi HotStart ReadyMix (2X)Kapa BiosystemsCatalog #KK2602
EvaGreen® Dye, 20x in WaterGold BiotechnologyCatalog #E-670
SYBR Gold (10000x)Thermo Fisher ScientificCatalog #S11494
Sodium acetate (3M) pH 5.5Thermo Fisher ScientificCatalog #AM9740
2-Propanol or Isopropanol for molecular biologyMerck MilliporeSigma (Sigma-Aldrich)Catalog #I9516-500mL
Ethanol Pure 200 proof for molecular biologyMerck MilliporeSigma (Sigma-Aldrich)Catalog #E7023-500mL
DNA Clean & Concentrator-5 (capped)Zymo ResearchCatalog #D4014
Tris-borate-EDTA buffer (10X)Thermo Fisher ScientificCatalog #15581044
DNA LoBind Tubes 2.0 mLEppendorfCatalog #30108078
DNA LoBind Tubes 5mLEppendorfCatalog #30108310
8-strip PCR tube without cap (0.2mL)VWR International (Avantor)Catalog #20170-002
8-strip PCR tube with individually attached bubble VWR International (Avantor)Catalog #53509-304
Corning polypropylene tube (15mL)Fisher ScientificCatalog #0553859B
Corning polypropylene tube (50mL)Fisher ScientificCatalog #0553868
Reservoir for 8 channel pipetters (25mL) individually wrappedCatalog #28-132
Millex-GP syringe filter unit (0.22 µm)Merck Millipore (EMD Millipore)Catalog #SLGP033RS
Nanosep 0.2 µm column PallCatalog #ODM02C35
CELLTRICS 30 µm strainer Fisher ScientificCatalog #NC9682496
Scalpel bladeIntegra BiosciencesCatalog #4-410
Dual-chambered counting slideBio-Rad LaboratoriesCatalog #145-0011
Equipments List:
- Eppendorf ThermoMixer C with Thermo Top (Eppendorf, cat. no.2231000574)
- IKA MS3 digital orbital shaker, with MS 1.32 tube insert (Coleparmer, cat. no. UX-04304-04)
- Tube revolver/rotator (Thermo Fisher Scientific, cat. no. 88881001)
- 0.2 mL PCR Strip / 1.5 mL Microfuge magnetic separator (Permagen Labware, cat. no. SKU: MSR1224B)
- MiSeq (Illumina)
- MiSeq reagent kit v2 (300 cycles) (Illumina, cat. no. MS-102-2002)
- MiSeq reagent kit v3 (150 cycles) (Illumina, cat. no. MS-102-3001)
- Qubit 3.0 fluorometer (Thermo Fisher Scientific, cat. no. Q33216)
- E1-ClipTip multichannel pipette, 12 channel, 0.5-12.5 µL, 1-30 µL and 2-125 µL (Fisher Scientific, cat. no. 14-387-972TI, 14-387-973TI and 14-387-974TI)
- ClipTip 384 12.5 µL, 30 µL and 125 µL multichannel pipette tip, racked, filter, sterile (Thermo Fisher Scientific, cat. no. 94420053, 94420103 and 94420153)
- Refrigerated centrifuge (Eppendorf)
- Bench top centrifuge (Eppendorf)
- mySPIN mini centrifuge (Thermo Fisher Scientific, cat. no. 75004061)
- CFX96 Touch deep well real-time PCR detection system (BIO-RAD)
- T100 Thermocycler (BIO-RAD, cat. no. 1861096)
- TC20 Automated cell counter (BIO-RAD, cat. no.1450102)
- Dual-chambered counting slide (BIO-RAD, cat. no. 145-0011)
- Eppendorf PCR cooler (Eppendorf, cat. no. 022510525)
- XCell SureLock mini-cell electrophoresis system (Thermo Fisher Scientific, cat. no. EI0001)
- UV transilluminator
Reagent setup
3h
40% (wt/vol) PEG 6000. Weigh 16.0 g of PEG 6000 in 50 mL tube. Add nuclease-free water and bring the total volume to 40 mL. Rotate the tube at room temperature until PEG 6000 completely dissolved. Spin down the tube at 200 g for 2 min, at room temperature to remove the tiny bubble. CRITICAL: 40% (wt/vol) of PEG 6000 is very viscous and difficult to filter through a 0.22 µm filter. We suggest preparing 40% PEG freshly before making GLR buffer. When PEG is diluted in 4x GLR buffer, it is easier to filter.
4x GLR buffer. To prepare 40 mL of 4x GLR buffer, Add 2.64 mL of nuclease-free water, 10.56 mL of 1 M Tris-HCl, pH 8.0, 0.8 mL of 1 M MgCl2 and 4 mL of 2 M KCl into 50 mL tube. Gently mix well by vortexing. Add 22 mL of 40% (wt/vol) PEG 6000 and gently mix well by vortexing. Filter through 0.22 µm into a new 50 mL tube and briefly spin the tube at room temperature for 30 s. Aliquot 1.8 mL into 2 mL tubes to minimize contamination from each use and store at 4 ºC.
10% (vol/vol) Triton X-100. Slowly aspirate 2.0 mL of Triton X-100 liquid with low retention pipette tip and slowly dispend into 18.0 mL nuclease-free water in 50 mL tube. Dissolve Triton X-100 by slowly rotate the tube until the solution is clear. Filter 10% Triton X-100 solution through 0.22 µm syringe filter into a new 50 mL tube and store at room temperature. CRITICAL: If it’s difficult to pipette Triton X-100 accurately as it’s a viscous liquid, we may warm it at 37 ºC before pipetting
10% (vol/vol) Tween 20. Tween 20 is very viscous liquid and difficult to pipette accurately. We convert the volume in cm3 into grams using the density of tween 20 at 25 ºC is 1.1 cm3. To prepare 20 mL of 10% (vol/vol) Tween 20, weigh 2.2 g of Tween 20 in 50 mL tube. Add 18.0 mL of nuclease-free water and invert or rotate the tube slowly at room temperature until Tween 20 is completely dissolved in water. Filter 10% Tween 20 solution through 0.22 µm syringe filter into a new 50 mL tube and store at room temperature.
25 mM dNTP mix. Mix 250 µL each of 100 mM dATP, dCTP, dGTP and dTTP in 1.5 mL tube. Mix well by vortexing and briefly spin the tube at room temperature for 5 s. Aliquot 250 µL into each of 1.5 mL tube and store at -20 ºC for a couple of years
2x Lysis buffer. To prepare 25 mL of 2x Lysis buffer, add 6.5 mL of nuclease-free water into 50 mL tube. Add 0.5 mL of 1 M Tris-HCl, pH 8.0, 2 mL of 5 M NaCl, 5 mL of 0.5 M EDTA and 11 mL of 10% (wt/vol) SDS. Gently mix and aliquot 1.8 mL into 2 mL tubes and store at room temperature.
1x B&W-T buffer. To prepare 40 mL of 1x B&W-T, add 31.56 mL of nuclease-free water into 50 mL tube. Add 200 µL of 1 M Tris-HCl, pH 8.0, 8 mL of 5 M NaCl, 40 µL of 0.5 M EDTA and 200 µL of 10% (vol/vol) Tween 20. Gently mix by vortexing and store at room temperature.
2x B&W buffer. To prepare 25 mL of 1x B&W, add 14.7 mL of nuclease-free water into 50 mL tube. Add 250 µL of 1 M Tris-HCl, pH 8.0, 10 mL of 5 M NaCl, and 50 µL of 0.5 M EDTA. Mix well by vortexing and store at room temperature.
0.1 M PMSF. Weigh 34.8 mg of PMSF and transfer into 2 mL microtube. Add 100% isopropanol to 2 mL and vortex vigorously to dissolve PMSF. Quick spin the tube down and transfer all solution into 3 mL syringe. Filter through a 0.22 µm syringe filter to a new 2 mL tube. Aliquot 50 µL per 0.2 mL PCR tube and store at -20 ºC for up to 4 months. CRITICAL: To maintain the activity of PMSF in solution, store PMSF in single-use aliquots.
Transposon preparation. Resuspend Nextera adapter 1, 5P-Nextera adapter 2 and mosaic end (ME) oligos with nuclease-free water to 100 µM. Mix 500 µL of 100 µM Nextera adapter 1 and 500 µL of 100 µM ME in 1.5 mL DNA LoBind microtube, aliquot 30 µL of non-annealed transposons into each of 0.2 mL PCR tube and store at -20 ºC for up to 1-2 years. Prepare 5P-Nextera adapter 2 and ME the same way as Nextera adapter 1. All sequences of oligos can be found in the attached tables.
Samples hashing barcodes preparation. Resuspend oligonucleotides barcodes with nuclease-free water to 10 pM final concentration and make 20 uL aliquots of each oligo in 0.2 mL PCR tubes.
| Oligo name | Sequence | |
| SNARE2-Truhash001 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTGAAGTGAGTTBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA | |
| SNARE2-Truhash002 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTCCTTCCGGTABAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA | |
| SNARE2-Truhash003 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTTCCGACGCATBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA | |
| SNARE2-Truhash004 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTAGGTAGCACABAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA | |
| SNARE2-Truhash005 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTGAATGCCGGTBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA | |
| SNARE2-Truhash006 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTCGTCAGGTCTBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA | |
| SNARE2-Truhash007 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTCGTACAGAGCBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA | |
| SNARE2-Truhash008 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTCTATATCGTCBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA | |
Hashing Oligos
Round 1 DNA barcoding plates generation
1h
Accessible chromatin (AC) Round 1 barcoded oligos preparation (Plate A). Prepare 50 µL of 25 µM accessible chromatin (AC) Round 1 barcoded oligos annealed with 23 µM accessible chromatin (AC) Round 1 linker oligos (BC_0100)
Resuspend AC Round 1 linker (BC_0100) with nuclease-free water to final concentration of 1 mM
Prepare 2.5 mL of 30.67 µM AC Round 1 linker by adding 76.68 µL of 1 mM of AC Round 1 linker to 2,423.32 µL of nuclease-free water in 5 mL tube and mix well by vortexing
Add 12.5 µL of 100 µM AC Round 1 barcoded oligos into 96-well plate (total 48 wells, rows A - D) with multichannel pipette
Transfer AC Round 1 linker oligos into 25 mL reservoir
Add 37.5 µL of 30.67 µM AC Round 1 linker into each well of AC Round 1 barcodes with multichannel pipette and mix well by pipetting 12 times (mixing volume 45 µL)
CRITICAL STEP: Seal and spin down the plate on 96-well plate swinging bucket rotor at 160 g for 1min, 4 ºC.
Anneal AC Round 1 barcoded oligos and AC linker oligos on thermocycler using the following annealing program: 95 ºC for 2 min, slowly cool down to 20 ºC (0.1 ºC/s) and hold at 4 ºC. CRITICAL STEP: After annealing oligonucleotides in the plate, we recommend spinning down the plate and use a sterile needle to punch the holes on the sealing film to release the pressure in every single well. Otherwise, the liquid inside the well will be pulled up to the top of the well when the film is unsealed, and this can lead to barcode cross-contamination. PAUSE POINT: If do not want to continue to mix these annealed oligos in the next step, store that plate at -20 ºC
RNA reverse transcription (RT) Round 1 barcoded oligos preparation (Plate B). Prepare 50 µL of 25 µM of oligo (dT)15 and 25 µM of random hexamer (N6) RT barcoded oligos (see attached tables) mix in each of 48 wells. CRITICAL STEP: 100 µM of dT and N6 reverse transcription barcoded oligos are ordered in row A-D and row E-H of oligo plate, respectively.
Transfer 12.5 µL of 100 µM of rows A-D (dT) in the RT barcoded oligos plate to rows A-D of a new 96-well DNA LoBind plate. CRITICAL STEP If use electronic multichannel pipette, add 1 µL of air after aspirating to avoid cross-contamination of barcoded oligos. This can be applied to other steps when we have to transfer barcoded oligos from stock plate to a new plate. Make sure that oligos are delivered to the bottom of the well.
Transfer 12.5 µL of 100 µM of rows E-H oligos (N6) in RT barcoded oligos plate to rows A-D (row E to row A, row F to row B, row G to row C, row H to row D) of 96-well plate that contains dT barcoded oligos above
Pipette 2 mL of nuclease-free water into 25 mL reservoir
Add 25 µL of nuclease-free water to row A-D of RNA Round 1 stock plate and mix well by pipetting 12 times (mixing volume 45 µL)
Seal and spin down the plate at 160 g for 1 min, 4 ºC and leave the plate on ice or PCR cool rack
AC Round 1 barcoded oligos and RT Round 1 barcoded oligos mix. The final concentration of each oligo (dT, N6, AC) in the oligo mix is 12.5 µM.
Transfer 50 µL of oligos in plate B (RT Round 1 barcoded oligos) into plate A that contain 50 µL of annealed AC Round 1 barcoded oligos and linker at identical well IDs (rows A-D) and mix well by pipetting 12 times (mixing volume 90 µL)
Spin down the plate at 160 g for 1 min, 4 ºC and put the plate on PCR cool rack
Aliquot 4 µL of mixed Round 1 barcoded oligos (rows A-D) into 10-25 of 96-well plates as “working plates”
Spin down working plates at 160 g for 1 min, 4 ºC and store at -20 ºC for up to a couple of years. Store the left over Round 1 stock plate at -20 ºC.
Round 2 DNA barcoding plates generation
1h
Ligation Round 2 barcoded oligos. Prepare stock plate of 100 µL of 18 µM ligation Round 2 barcoded oligos annealed with 16.5 µM ligation Round 2 linker (BC_0215).
Resuspend ligation Round 2 linker (BC_0215) with nuclease-free water to final concentration of 1 mM
Prepare 9 mL of 20.12 µM ligation Round 2 linker by adding 181.08 µL of 1 mM round 2 linker to 8,818.9 µL of nuclease-free water in 15 mL tube and mix well by vortexing
Add 18 µL of 100 µM of ligation round 2 barcoded oligos into 96-well plate (rows A-H)
Transfer ligation Round 2 linker into 25 mL reservoir
Add 82 µL of 20.12 µM ligation Round 2 linker to each well of ligation Round 2 barcoded oligos with and mix well by pipetting 10 times (mixing volume 90 µL)
Seal and spin down the plate at 160 g for 1min, 4 ºC
Anneal ligation Round 2 barcoded oligos and Round 2 linker on thermocycler using the annealing program for Round 1 barcoded oligos and keep on ice
Spin down the plate at 160 g for 1min, 4 ºC and keep on ice
Aliquot 10 µL of annealed ligation Round 2 barcoded/linker oligos into 10 of 96-well plate as “working plate”
Spin down the working plate at 160 g for 1min, 4 ºC before store at -20ºC for up to a couple of year
Round 3 DNA barcoding plates generation
1h
Ligation Round 3 barcoded oligos. Prepare stock plate of 100 µL of 21 µM Round 3 barcoded oligos annealed with 19.5 µM ligation Round 3 linker (BC_0060).
Resuspend ligation Round 3 linker (BC_0060) with nuclease-free water to final concentration of 1 mM
Prepare 8.5 mL of 24.68 µM ligation Round 3 linker by adding 209.8 µL of 1 mM Round 3 linker to 8,290.2 µL of nuclease-free water in 15 mL tube and mix well by vortexing
Add 21 µL of 100 µM of ligation Round 3 barcoded oligos into 96-well plate (rows A-H)
Transfer ligation Round 3 linker into 25 mL reservoir
Add 79 µL of 24.68 µM ligation Round 3 linker to each well of ligation Round 3 barcoded oligos and mix well by pipetting 12 times (mixing volume 90 µL)
Seal and spin down the plate at 160 g for 1min, 4 ºC
Anneal ligation Round 3 barcoded oligos and Round 3 linker on thermocycler using the annealing program for Round 1 barcoded oligos and keep on ice
Spin down the plate at 160 g for 1min, 4 ºC and keep on ice
Aliquot 10 µL of annealed ligation Round 3 barcoded/linker oligos into 10 of 96-well plate as “working plate”
Spin down the working plate at 160 g for 1min, 4 ºC before store at -20ºC for up to a couple of year
Transposase preparation
2h
Note
Convert Tn5 transposase (Diagenode Tagmentase, Diagenode #C01070010-20, EZ-Tn5 Transposase, Lucigen #TNP92110, or expressed/purified with an in house protocol). For example, if the concentration of unloaded Tn5 is 0.40 mg/mL, the monomer concentration in uM is 7.55 µM based on molar mass of monomer Tn5 ~53,000 g/mol [0.4 mg/mL * (1 mol/53,000 g)]. CRITICAL STEP: Anneal transposons and load Tn5 on the day of experiment 1-2 h prior experiment starts. Combine transposons and mix well before adding Tn5 to make sure that both transposons are sufficiently homogeneous prior to mixing with Tn5. Avoid generating bubbles when mixing Tn5 with transposons by pipetting slowly and do not fill pipette tip with air. If the experiment is not ready, store loaded Tn5 at -20 ºC.
Thaw non-annealed transposons (Nextera adapter 1/ME and 5P-Nextera adapter 2/ME) on ice. Briefly vortex and quick spin the tube. Anneal transposons on thermocycler using following program: 95 ºC 5 min, slowly cool down to 65 ºC (0.1 ºC/s), 65 ºC 5 min, slowly cool down to 4 ºC (0.1 ºC/s) and hold at 4 ºC.
Load transposons 1.5x molar ratio to Tn5. The amount below is sufficient for tagmenting ~ 1.8 million nuclei/cells.
13 µL 37.72 uM monomer Tagmentase (2mg/mL)
7.35 µL 50 uM Annealed Nextera adapter 1/ME
7.35 µL 50 uM Annealed 5P-Nextera adapter 2/ME
39 µL Tagmentase Dilution Buffer
Add annealed Nextera adapter 1/ME and annealed 5P-Nextera adapter 2/ME into the bottom of 1.5 mL DNA LoBind microtube, mix well by pipetting 10 times or gently vortexing and briefly spin the tube on mini centrifuge for 3 s. Addunloaded Tn5 and mix by gently pipetting 20 times (set the volume of p200 pipette to 80 µL). Quick spin the tube and incubate at 25 ºC for 0.5 h, 350 rpm. The final concentration of loaded Tn5 is 7.35 µM (monomer Tn5 concentration).
00:30:00
30m
Nuclei isolation and fixation
3h
Isolate nuclei from tissue following tissue-specific nuclei extraction protocol (dx.doi.org/10.17504/protocols.io.ufketkw) with 0.1 U/µL of SUPERase In RNase Inhibitor and 0.2 U/µL of Enzymatics RNase Inhibitor. For cell lines, nuclei can be extracted with ATAC Lysis buffer with 0.1% NP-40 as previously described with the addition of RNase inhibitors, and increase lysis volume proportional to the number of input cells.
To each tube of 0.5 mL volumes of nuclei suspensions, add 15 µL of hashing oligos. Make a note of which oligo was added to which sample. Mix briefly, centrifuge at 500 g for 8 minutes at 4C, remove 400 µL of supernatant. Then pool up to 8 barcoded samples
Prepare 1x PBS + RI (1 mL per sample) and keep on ice.
1000 µL PBS, pH 7.4
2.5 µL SUPERase In (20 U/uL)
1.25 µL Enzymatics Rnase In (40 U/uL)
Prepare 1% (wt/vol) formaldehyde in 1x PBS (1 mL per sample) and keep on ice. CRITICAL: formaldehyde solution should be in 1x PBS and methanol free.
937.5 µL PBS, pH 7.4
62.5 µL Formaldehyde, 16% wt/vol
Resuspend 1-2 million nuclei with 1 mL 1x PBS + RI and keep on ice. To each tube of 1 mL volumes of nuclei suspensions, add 15 µL of hashing oligos. Make a note of which oligo was added to which sample. Mix briefly.
Add 1 mL of 1% formaldehyde to nuclei suspension and pipette gently 8 times. Leave the tube on ice for 10 minutes.
00:10:00 Fixation
Pellet nuclei at 900 g for 8 min at 4C using bucket rotor centrifuge.
Prepare 1x PBS + 0.1% (wt/vol) BSA + RI ( 1 mL per sample) and leave on ice.
1000 µL PBS, pH 7.4
13.4 µL BSA, 7.5% wt/vol
1.5 µL SUPERase In (20 U/uL)
0.75 µL Enzymatics RNase In (40 U/uL)
Prepare 1x Tango Buffer + RI (1 mL per ~ 3 million nuclei/cells) and leave on ice.
100 µL Tango Buffer, 10x
160 µL DMF, 100%
5 µL SUPERase In (20U/uL)
2.5 µL Enzymatics RNase In (40 U/uL)
732.5 µL Nuclease-free water
Aspirate the supernatant, resuspend pelleted nuclei with 0.1 mL of 1x PBS + 0.1% BSA + RI to wash. Pool up to 8 samples, ~800 uL total max. Add wash buffer to 1 mL total volume.
Pellet nuclei at 900 g for 8 min at 4C using bucket rotor centrifuge.
Aspirate the supernatant and resuspend with 1x Tango Buffer + RI to have a minimum concentration of 3,400 nuclei per microliter.
Count the nuclei using cell counter and resuspend the nuclei solution with additional 1x Tango Buffer + RI to obtain 3,400 nuclei per microliter.
Tagmentation
1h
Set up tagmentation mix per reaction as follows (1 reaction per 150,000 nuclei/cells). Minimum 4 reactions for 1 sample per plate (48 wells of round 1).
3 µL Tango Buffer, 10x
4.8 µL DMF, 100%
4.95 µL Loaded Tn5, 7.35uM
1.5 µL SUPERase In (20 U/uL)
0.75 µL Enzymatics RNase In (40 U/uL)
15 µL Nuclease-free water
Prepare 150,000 nuclei in 45 µL of 1x Tango Buffer and mix with 30 µL of tagmentation mix. The final concentration of Tn5 and DMF in final reaction is 0.8 µM and 16% (vol/vol), respectively. The ratio of nuclei suspension and tagmentation mix is 3 : 2 or 45 µL : 30 µL. Add nuclei into 1.5 mL DNA LoBind tube then add tagmentation mix and mix gently by pipetting 10 times. Briefly spin the tube on mini centrifuge at room temperature for 3 s and aliquot 75 µL of tagmentation reaction into 1.5 mL DNA LoBind tube.
CRITICAL STEP: Set up 4 tubes of tagmentation reactions to have enough nuclei for Round 1 barcoding (8,000 nuclei/well x total 48 wells = 384,000 nuclei). Set up the reactions on ice. Do not incubate tagmentation reaction in large volume to make sure that nuclei are distributed evenly in the reaction not sitting on the bottom of the tube when incubating during tagmentation.
Place the tubes on thermomixer and incubate at 37 ºC for 30 min, 300 rpm.
00:30:00 Tagmentation
Before incubation is complete, prepare 1x PBS + 0.1% (wt/vol) BSA + RI (1 mL per 400 µL tagmented nuclei) and keep on ice
1000 µL PBS, pH 7.4
13.33 µL BSA, 7.5% wt/vol
1.5 µL SUPERase In (20 U/uL)
0.75 µL Enzymatics RNase In (40 U/uL)
Remove the tubes from thermomixer and place on ice. Then pool tagmented nuclei of the same sample into the same tube.
Add 2.5x volume of 1x PBS + 0.1% (wt/vol) BSA + RI (1000 µL to 400 µL tagmented nuclei) to pooled tagmented nuclei and mix by pipetting gently 5 times and centrifuge at 900 g for 8 min, 4 ºC with swinging bucket rotor
During centrifugation, prepare 0.5x PBS + RI (1 mL per 1 million nuclei/cells) and keep on ice
500 µL PBS, pH 7.4
500 µL Nuclease free water
2.5 µL SUPERase In (20 U/uL)
1.25 µL Enzymatics Rnase In (40 U/uL)
CRITICAL STEP Aspirate supernatant and resuspend nuclei with 300 µL of 0.5x PBS + RI to have a minimum concentration of nuclei not lower than 1,000 nuclei/µL. If different numbers of nuclei in tagmentation are used, adjust suspension volume. Pipetette gently to resuspend
Count nuclei concentration with cell counter and dilute nuclei to 1,000 nuclei/µL with 0.5x PBS + RI.
Accessible chromatin (AC) oligo ligation
45m
Thaw Round 1 AC/RT oligo working plate on ice and spin the plate on swinging bucket at 200 g for 1 min, 4 ºC and leave the plate on ice. Note: Can leave in 4C at the beginning of Day 1 to thaw.
Prepare GLR-A mix following table below. CRITICAL STEP: Prepare GLR-A mix during washing tagmented nuclei, but add ATP, RNase inhibitor and T7 DNA Ligase just before ready to load into Round 1 barcoding plate
260 µL GLR Buffer, 4x
20.8 µL ATP, 100mM
52 µL dNTPs, 25 mM each
13 µL SUPERase In (20U/uL)
6.76 µL Enzymatics RNase In, (40U/uL)
10.4 µL DTT, 1M
78 µL T7 DNA Ligase (3000U/uL)
1.04 µL Nuclease-free water
Add 8 µL of nuclei to each well (row A – D) with a multichannel pipette.
Aliquot 73 µL of GLR-A into 6 tube-strip on PCR cool rack and add 8.5 µL of GLR-A mix to each well with a multichannel pipette.
Seal and quick spin the plate at 160 g for 15 s, 4 ºC. Gently mix reaction mix, Round 1 barcoded oligos and nuclei 5 times (mixing volume 18 µL)
Seal and quick spin the plate at 160 g for 10 s, 4 ºC and incubate the plate on thermomixer at 25 ºC for 30 min, 300 rpm
00:30:00 AC oligo ligation
Reverse transcription
45m
Aliquot 20 µL of Maxima H Minus Reverse Transcriptase into 6 tubes of PCR strip tube
120 µL Maxima H Minus RT (200 U/uL)
Remove Round 1 barcoding plate from thermomixer, put on PCR cool rack, and add 2.2 µL of Maxima H Minus Reverse Transcriptase to each well with a multichannel pipette. CRITICAL STEP: To get accurate volumes, set the speed of multichannel pipette to be very slow for aspirating and dispensing as the enzyme is very viscous
Seal and quick spin the plate at 160 g for 15 s, 4 ºC to bring enzyme to the bottom of the well and mix by gently pipetting 5 times (mixing volume 18 µL). Seal and quick spin the plate at 160 g for 10 s, 4ºC
Incubate the plate on thermocycler using the program: 50ºC for 10 min, 3 cycles of (8ºC for 12 s, 15ºC for 45 s, 20ºC for 45 s, 30ºC 30 s, 42ºC for 2 min, 50ºC for 3 min), 50ºC for 5 min.
Round 2 DNA barcoding
1h 30m
Before reverse transcription finishes, prepare 3 mL of 1x PBS + 0.1% BSA + RI
3000 µL PBS, pH 7.4
40 µL BSA, 7.5% wt/vol
4.5 µL SUPERase In (20 U/uL)
2.25 µL Enzymatics RNase In (40 U/uL)
Remove the plate from thermocycler and put on ice or PCR cool rack then pool all reactions into a chilled 25 mL reservoir and transfer pooled reaction into a chilled 5 mL DNA LoBind tube. CRITICAL STEP: Before pooling, pipette Round 1 barcoding plate 2 times to kick up nuclei from the bottom of the well and make sure that you transfer all reactions from the well by aspirating slowly and set pipette volume to 26 µL. This technique should be done for all pooling steps. Also keep reactions on ice all the time when handling samples or reaction mixes to prevent RNA degradation and preserve enzyme activity or temperature sensitive reagents like ATP.
Add 2.8 mL of 1x PBS + 0.1% (wt/vol) BSA + RI (2.5x volume) to rinse the basin and transfer buffer to the tube
Add 19.5 µL of 10% (vol/vol) Triton X-100 (final concentration of Triton X-100 is 0.05% (vol/vol)) and mix by inverting the tube 5 times before centrifuge at 900 g for 8 min, 4 ºC
19.5 µL Triton X-100, 10% vol/vol
Thaw Round 2 barcoded oligos working plate on ice and spin the plate at 200 g, for 1 min, 4 ºC and leave the plate on ice. Note: Can leave in 4C at the beginning of Day 1 to thaw.
Prepare 1x Buffer 3.1 as follows:
210 µL NEBuffer 3.1, 10x
21 µL Enzymatics RNase In (40U/uL)
1890 µL Nuclease free water
Prepare Ligation Mix as follows:
510 µL T4 DNA Ligase Buffer, 10x
40.8 µL Enzymatics RNase In (40U/uL)
12.75 µL SUPERase In (20U/uL)
51 µL BSA (20 mg/mL)
127.5 µL T4 DNA Ligase (400U/uL)
1338.8 µL Nuclease free water
Remove supernatant as much as possible (~20 µL left) and resuspend nuclei with 2.02 mL of 1x Buffer 3.1 and add 2.04 mL of Ligation mix and mix by pipetting 10 times
Transfer nuclei in ligation mix into 25 mL reservoir and add 40 µL of nuclei suspension into each well of Round 2 barcoding plate and mix gently by pipetting 5 times (mixing volume 45 µL)
Seal the plate with sealing film and quick spin the plate at 160 g for 10 s, 4 ºC
Incubate the plate on thermomixer at 37 ºC for 30 min, 300 rpm
00:30:00 Round 2 barcoding
Prepare Round 2 blocking solution as follows:
47.52 µL BC_0216, 1000 uM
300 µL T4 DNA Ligase Buffer, 10x
852.5 µL Nuclease free water
Remove Round 2 DNA barcoding plate from thermomixer and quick spin at 160 g for 10 s, 4 ºC. Add 10 µL of Round 2 blocking solution to each well with multichannel pipette and mix by pipetting gently 5 times (mixing volume 55 µL)
Seal the plate with sealing film and quick spin at 160 g for 10 s, 4ºC then incubate the plate on thermomixer at 37 ºC for 30 min, 300 rpm
00:30:00 Round 2 blocking
Round 3 DNA barcoding
1h
Thaw Round 3 barcoded oligos working plate on ice, spin the plate on swinging bucket at 200 g for 1 min, 4 ºC and leave the plate on ice
Place Round 2 DNA barcoding plate on PCR cool rack and pool into 25 mL reservoir. Add 100 µL of T4 DNA Ligase (400 U/µL) into the basin with nuclei from Round 2 barcoding plate and mix well by gently pipetting 10 times and rock the basin from side-to-side 10 times
100 µL T4 DNA Ligase, (400 U/uL)
Add 50 µL of nuclei suspension to each well of Round 3 DNA barcoding plate and mix gently by pipetting 5 times (mixing volume 55 µL).
Seal the plate with sealing film and briefly spin the plate at 160 g for 10 s, 4ºC. Incubate the plate on thermomixer at 37 ºC for 30min, 300 rpm
00:30:00 Round 3 barcoding
Prepare Round 3 blocking solution as follows:
41.4 µL BC_0066, 1000 uM
600 µL EDTA, 500mM
1758.6 µL Nuclease free water
Remove Round 3 DNA barcoding plate from thermomixer and add 20 µL of Round 3 blocking solution to each well and gently mix by pipetting 3 times (mixing volume 75 µL)
Without incubation, pool the reaction into 25 mL reservoir placed on ice, transfer supernatant into 15 mL tube and centrifuge at 1,000 g for 8 min, 4ºC. OPTIONAL: EDTA in the reaction inhibits ligase activity, therefor there is no need to change the pipette tips when pooling Round 3 ligation reaction. Set pipetting volume 85 µL when pooling nuclei.
In parallel, prepare chilled wash buffer as follows.
4000 µL PBS, pH 7.4
40 µL Triton X-100, 10% vol/vol
10 µL SUPERase In (20U/uL)
Also in parallel, thaw 2X Lysis buffer at 37C for ~15 minutes if previously prepped. If not see step 6 for recipe.
Remove supernatant and add 4 mL of wash buffer. Pipette gently 5 times with p1000 pipette then centrifuge at 1,000 g for 8 min, 4 ºC
Carefully remove supernatant as much as possible and resuspend nuclei with 300 µL of 1x PBS + RI
500 µL PBS, pH 7.4
5 µL SUPERase In (20 U/uL)
2.5 µL Enzymatics Rnase In (40 U/uL)
Count nuclei concentration with cell counter and aliquot nuclei at required number in each pool in 1.5 mL DNA LoBind tube and adjust total volume of nuclei to 50 µL with 1x PBS + RI. CRITICAL STEP: To sequence all nuclei, recommend using ≤20,000 cells per pool by splitting 50 µL of nuclei suspension into 6 - 12 tubes per plate.
Nuclei lysis
2h
Add 50 µL of 2x Lysis buffer and 10 µL of 20 mg/mL Proteinase K to each pool, mix well by gently vortexing and brief spin the tube down at room temperature for 10 s to collect all nuclei suspension to the bottom of the tube and incubate on thermomixer at 55 ºC for 2 h, 350 rpm to lyse nuclei and reverse crosslink formaldehyde fixation.
02:00:00 Nuclei lysis
Put nuclei lysate at -80 ºC to inactivate Proteinase K before continue to day 2 experiment. CRITICAL STEP: Lysis buffer tends to precipitate at room temperature or low temperature. Redissolve by incubating the tube at 37 ºC until it completely dissolves before adding to nuclei suspension. PAUSE POINT: Nuclear or cell lysate can be store at -80 ºC for up to 6 months before continue to day 2 experiment.
Dynabeads MyOne streptavidin beads preparation
20m
Prepare 3.5 mL of 1x B&W-T + RI for bead washing:
3500 µL B&W-T, 1x
5 µL SUPERase In (20U/uL)
Vortex the bottle of Dynabeads MyOne C1 thoroughly and aliquot required volume (44 µL/pool * number of pool) into 1.5 mL tube. Add 800 µL of 1x B&W-T + RI, mix by vortexing and pulse spin on mini centrifuge at room temperature for 3 s
44 µL MyOne C1 beads
Place the tubes onto the magnetic rack until liquid is clear; Remove supernatant with p1000 pipette
Resuspend the beads with 800 µL of 1x B&W-T + RI, vortex and pulse spin on mini centrifuge at room temperature for 3 s
Place the tubes onto the magnetic rack until solution is clear and remove supernatant with p1000 pipette
Repeat washes two more times (total of 3 washes)
Resuspend the beads with 100 µL of 2x B&W + RI per pool:
100 µL 2x B&W-T
2 µL SUPERase In (20U/uL)
cDNA/DNA capture
1h 30m
During bead preparation, take the tubes of lysate out of -80 ºC and place onto thermomixer set at 55 ºC for 2 min until lysate is completely thawed
Add 5 µL of 0.1 M PMSF (from -20 ºC) to each tube, pulse vortex for 10 s and pulse spin on mini centrifuge at room temperature for 5 s and incubate at room temperature for 10 min with no shaking
5 µL PMSF, 100 mM
00:10:00
Add 100 µL of streptavidin beads in 2x B&W + RI to each tube of lysate (no pipetting required) then agitate the tubes on mixer at room temperature for 1 h, 1,200 rpm. CRITICAL STEP: We recommend to use orbital shaker for microtubes. The speed of mixer can be adjusted as long as the beads do not settle on the bottom of the tube
01:00:00 Binding to beads
Pulse spin the tubes on mini centrifuge at room temperature for 5 s, place onto magnetic rack until solution is clear and remove supernatant with p200 pipette. CRITICAL STEP: Every time before placing the tube back to the magnet, quick spin the tubes on mini centrifuge for 3-5 s to collect all supernatant/lysate/buffer and beads to the bottom of the tube. Use p200 pipette to remove supernatant to avoid disturbing the beads and prevent bead loss.
Prepare 1 mL of 1x B&W-T + RI (750 uL per lysate) for bead washing as follows:
1000 µL B&W-T, 1x
1.4 µL SUPERase In (20U/uL)
Add 250 µL of 1 x B&W-T + RI and agitate the bead at room temperature for 5 min, 1,500 rpm to wash the beads
00:05:00 wash with 1x B&W-T + RI
Place the tube onto magnetic rack until solution is clear and remove supernatant. Continue to template switching oligo blocking on AC DNA immediately.
Blocking template switching oligo
30m
During working on Steps 109-110, prepare Nextera adapter 1 blocking solution per pool:
250 µL SSC, 6x
2.5 µL Nextera adapter 1 blocker, 100 uM
1 µL SUPERase In (20U/uL)
Add 250 µL of 6x SSC to each tube without bead suspension and wait until the supernatant is clear then remove supernatant with p200 pipette
250 µL SSC, 6x
Add 250 µL of Nextera adapter 1 blocking solution to each tube and agitate the tubes on mixer at room temperature for 1 min, 1,500 rpm then reduce the speed to 500 rpm for 14 min. CRITICAL STEP: Agitate the tubes at high speed 1,500 rpm for 1 min to make sure the beads are resuspended well, then shake gently at 500 rpm for 14 min to allow hybridization of Nextera adapter 1 blocker and Nextera adpter 1 on AC DNA and make sure that the beads do not settle.
00:15:00 incubate with Nextera Blocking Solution
Place the tube onto magnetic rack until solution is clear and remove supernatant with p200 pipette.
Wash the beads twice with 1x B&W-T + RI as described above at room temperature for 5 min each round.
00:05:00 wash with 1x B&W-T + RI
00:05:00 wash #2 with 1x B&W-T + RI
In parallel, prepare Tris-T + RI during second bead wash
250 µL Tris-HCl, pH 8.0, 10 mM
2.5 µL Tween 20, 10% vol/vol
0.63 µL SUPERase In (20U/uL)
Wash the beads with 250 µL of Tris-T + RI (5 min) the same way as 1x B&W-T + RI wash (at room temperature for 5 min, 1,500 rpm). In parallel, prepare GLR-B mix. CRITICAL STEP: If GLR-B mix is not ready, leave the beads in the tube with Tris-T + RI on ice until GLR-B mix is ready
00:05:00 wash with Tris-T + RI
Gap filling, ligation and complete reverse transcription
2h 15m
During washing, prepare GLR-B mix as follows:
50 µL GLR buffer, 4x
4 µL ATP, 100mM
20 µL Ficoll PM 400, 20% wt/vol
10 µL dNTPs, 25 mM each
5 µL SUPERase In (20U/uL)
2 µL DTT , 1M
12.5 µL Hemo Klentag
2.5 µL T7 DNA Ligase (3000U/uL)
89 µL Nuclease free water
Place the tubes onto magnetic rack until solution is clear, remove supernatant with p200 pipette and add 250 µL of nuclease-free water to each tube without bead suspension
250 µL Nuclease free water
Remove water and resuspend the beads with 195 µL of GLR-B mix by gently vortexing and quick spin the tubes on mini centrifuge for 3 s
Rotate the tubes in incubator at 37 ºC with slow speed for 15 min to allow for gap filling on AC DNA and ligate Nextera adapter 1 blocker to the AC DNA. OPTIONAL: For any step required rotator, thermomixer can be alternatively used as long as the tube is shaken gently and the beads do not settle.
00:15:00 Gap filling and ligation
Remove the tubes from incubator and add each tube with 5 µL of 100 µM TSO and 5 µL of Maxima H Minus reverse transcriptase and mix well by gently vortexing.
5 µL TSO oligo, 100 uM
5 µL Maxima H Minus RT, (200 U/uL)
Continue to incubate at room temperature for 30 min with slow rotation.
00:30:00 complete reverse transcription
Incubate at 42 ºC for 90 min with slow rotation.
01:30:00 complete reverse transcription
PAUSE POINT: The beads can be stored in Tris-T buffer at 4 ºC overnight before continuing to 1st PCR (Remove supernatant and replace with Tris-T buffer). However, we recommend to continue to the 1st PCR immediately if possible.
First PCR, cDNA/DNA amplification
1h 30m
Before finishing 42C incubation, set up the first PCR mix to amplify both cDNA and accessible chromatin (AC) DNA and prepare PCR strip tubes with individual hinged cap (4 tubes per pool):
110 µL KAPA HiFi HotStart ReadyMix, 2x
6.9 µL BC_0108, 10 uM
17.6 µL BC_0062, 10 uM
6.9 µL BC_0082, 10 uM
6.9 µL Hash_PCR, 10 uM
74.8 µL Nuclease free water
Note:
Sequence of Hash_PCR: ACACTCTTTCCCTACACGACGCTCTTCCGATCT
After finishing incubation at 42 ºC for 90 min, place the tubes onto the magnetic rack until liquid is clear and remove supernatant with p200 pipette
Add 250 µL of nuclease-free water to each tube without bead resuspension. Once liquid is clear, remove supernatant.
250 µL Nuclease free water
Resuspend the beads with 220 µL of first PCR mix, quick spin and aliquot 55 µL of bead suspension in PCR mix to each of 4 PCR strip tubes. Transfer all the leftover beads to 4 PCR tubes equally. CRITICAL STEP: Transfer the beads in PCR mix directly to the bottom of the tube, so there is no need to spin the tube before placing on thermocycler. Try to transfer PCR mix with the beads into PCR strip tubes as quick as possible to minimize bead settling before the reaction starts.
Place the tubes on thermocycler and run following program: 95 ºC for 3 min, 9 cycles of (98 ºC for 20 s, 58 ºC for 45 s, 72 ºC for 3 min), 72 ºC for 5 min, 4 ºC hold.
PAUSE POINT PCR reaction can be stored at -20 ºC for a month or 4 ºC for a week.
Place strip tubes onto 0.2 mL magnetic rack until supernatant is clear and pool 1st round PCR products from 4 strip tubes of the same pool into 1.5 mL DNA LoBind tube
Vortex the tube and quick spin on mini centrifuge for 3 s before splitting PCR products into two parts (100 µL each), “AC” for chromatin accessibility (AC) library preparation and “R” for RNA library preparation. Note: AC and R in this step are the same PCR products but will be bead size-selected at different bead volume ratio.
sn/scATAC libraries: purifiation and validation
1h
Perform one round of 1.2x KAPA Pure Beads purification following manufacturer’s instructions by using 120 µL of KAPA Pure Beads with 100 µL of PCR products and elute with 40 µL of nuclease-free water
1. Binding
120 µL KAPA Pure Beads 00:08:00
2. Washing
180 µL Ethanol, 100% 00:00:30
3. 2nd wash
180 µL Ethanol, 100% 00:00:30
4. Drying
00:01:00 37 °C
5. Elution
40 µL Nuclease free water 00:10:00 37 °C
Transfer eluent into new PCR strip tubes. The resultant is called AC-A.
Use 2 µL of AC-A product to determine DNA concentration with Qubit dsDNA HS assay kit following manufacturer’s instruction
Verify ~10 ng of AC-A in 6% TBE gel and run in 1x TBE buffer at 250 volts for 23 min with 0.5 µL of Low DNA Mass Ladder as reference
sn/scATAC libraries: 2nd PCR and library preparation
6h 30m
In PCR strip tubes, use 5 ng of AC-A as template for enrichment of AC DNA over cDNA and adjust volume of template to 10 µL with nuclease-free water and quick spin down the tubes. CRITICAL STEP: If the concentration of PCR product is higher than 5 ng/µL, it tends to have high error to pipette the volume smaller than 1 µL. We recommend to aliquot AC-A, dilute into 0.5 ng/µL in total 30-50 µL and use 10 µL as template for AC 2nd PCR.
10 µL AC-A DNA (total 5 ng)
Prepare PCR mix as follows:
25 µL KAPA HiFi HotStart ReadyMix, 2x
2.5 µL SPLiT_N701, 10 uM
2.5 µL EvaGreen, 20x
7.5 µL Nuclease free water
Add 37.5 µL of PCR mix and 2.5 µL of 10 µM Ad1_N50X (attached tables) into the tube with AC-A template
2.5 µL Ad1_N5XX, 10 uM
Mix by gently vortexing, quick spin PCR tubes on mini centrifuge at room temperature for 3 s and run qPCR on real-time PCR machine: 95 ºC for 3 min, 12 (or fewer) cycles of (98 ºC for 20 s, 58 ºC for 45 s, 72 ºC for 1 min), 72 ºC for 5 min, 4 ºC hold.
PAUSE POINT: PCR reaction can be stored at -20 ºC for a month or 4 ºC for a week.
Purify PCR product with DNA Clean & Concentrator following manufacturer's instructions and elute with 40 µL of DNA Elution Buffer. Resultant is called AC-B
Use 2 µL of AC-B to determine DNA concentration with Qubit dsDNA HS assay kit following manufacturing’s instruction
Verify ~10 ng of AC-B in 6% TBE gel and run in 1x TBE buffer at 250 volts for 23 min with 0.5 µL of Low DNA Mass Ladder as a reference in separate lane. CRITICAL STEP: We expect to see nucleosome pattern with larger size (~125 bp larger) compare to standard ATAC-seq due to the presence of cell barcodes and linker sequences
Pool equimolar ratio of AC-B libraries (~200-250 ng/pool) and perform PAGE size-selection at the size between 300-1,000 bp.
Use 2 µL of AC sequencing libraries to determine for DNA concentration with Qubit dsDNA HS assay
Verify ~5 -10 ng of AC sequencing libraries in 6% TBE gel and run in 1x TBE buffer at 250 volts for 23 min or by TapeStation
sn/scATAC libraries: Quality validation by MiSeq sequencing
1d
Validate AC sequencing libraries with MiSeq sequencing using v2 reagent kit by loading at 20 pm based on Qubit dsDNA HS quantification with at least 5% PhiX spike in following Illumina loading guide. The sequencing configuration is 75 cycles for read 1, 94 cycles for index 1, 8 cycles for index 2, and 75 cycles for read 2.
Note
CRITICAL STEP: For MiSeq sequencing, quantification of sequencing library concentration by Qubit and determination of average library size is sufficient. For high-throughput sequencing, we recommend to run sequencing libraries on Bioanalyzer, TapeStation or equivalent instrument that can determine accurate average library size and contamination of adapter dimers peaks at approximately 125-175 bp. We recommend to run qPCR to quantify sequencing library concentration using the average library size derived from TapeStation. For a two-channel sequencing system such as NovaSeq, we recommend to spike-in with minimum of 10% PhiX or consult sequencing core.
Mix 1:1 ratio of "SN2-AC R1" sequencing primer pairs for sequencing read 1 on Illumina workflow A (HiSeq 2500, MiSeq, NovaSeq 6000 platforms).
3 µL SNARE2_Read1, 100 uM
3 µL PhiX_Read1, 100 uM
594 µL Hybridization buffer, HT1
Mix 1:1 ratio of "SN2-AC R2" sequencing primer pairs for sequencing index 1 on Illumina workflow A (HiSeq 2500, MiSeq, NovaSeq 6000 platforms).
3 µL SNARE2-AC_BCread, 100 uM
3 µL PhiX_Read1, 100 uM
594 µL Hybridization buffer, HT1
Mix 1:1 ratio of "SN2-AC R4" sequencing primer pairs for sequencing read 2 on Illumina workflow A (HiSeq 2500, MiSeq, NovaSeq 6000 platforms).
3 µL SNARE2-AC_Read2, 100 uM
3 µL PhiX_Read2, 100 uM
594 µL Hybridization buffer, HT1
sn/scRNA libraries: purification and validation
1h
Perform 1 round of 0.8x KAPA Pure Beads purification following manufacturer’s instructions by mixing 100 µL of 1st PCR product + with 80 µL of KAPA Pure Beads and elute with 40 µL of nuclease-free water. Resultant is called R-A.
1. Binding
80 µL KAPA Pure Beads 00:08:00
2. Washing
180 µL Ethanol, 100% 00:00:30
3. 2nd wash
180 µL Ethanol, 100% 00:00:30
4. Drying
00:01:00 37 °C
5. Elution
40 µL Nuclease free water 00:10:00
Use 2 µL of R-A to determine DNA concentration with Qubit dsDNA HS assay following manufacturer’s instructions
Verify 5-10 ng of R-A in 6% TBE gel and run in 1x TBE buffer at 250 volts for 23 min with 0.5 µL of Low DNA Mass Ladder as a reference
sn/scRNA libraries: 2nd PCR, purification, and validation
2h
In PCR strip tubes, use 5 ng of R-A as template for enrichment of cDNA over AC DNA and adjust the volume of template to 10 µL with nuclease-free water and quick spin down the tubes. CRITICAL STEP: If the concentration of PCR product is higher than 5 ng/µL, it tends to have high error to pipette the volume smaller than 1 µL. We recommend to aliquot R-A, dilute into 0.5 ng/µL in total 30-50 µL and use 10 µL as template for RNA 2nd PCR
Prepare PCR mix for second round amplification of cDNA as follows:
25 µL KAPA HiFi HotStart ReadyMix, 2x
2 µL BC_0108, 10 uM
2 µL BC_0062, 10 uM
2.5 µL EvaGreen, 20x
8.5 µL Nuclease free water
Add 40 µL of PCR mix to each tube, gently vortex and quick spin PCR tubes on mini centrifuge at room temperature for 3 s
Run the reactions on real-time PCR using following program: 95 ºC for 3 min, 12 (or fewer) cycles of (98 ºC for 20 s, 67 ºC for 45 s, 72 ºC for 3 min), 72 ºC for 5 min, 4 ºC hold.
PAUSE POINT PCR reaction can be stored at -20 ºC for a month or 4 ºC for a week.
Purify PCR products with 1 round of 0.8x KAPA Pure Beads and elute with 40 µL of nuclease-free water. The resultant purified DNA is called R-B
1. Binding
40 µL KAPA Pure Beads 00:08:00
2. Washing
180 µL Ethanol, 100% 00:00:30
3. 2nd wash
180 µL Ethanol, 100% 00:00:30
4. Drying
00:01:00 37 °C
5. Elution
40 µL Nuclease free water 00:10:00 37 °C
Determine DNA concentration with Qubit dsDNA HS assay using 2 µL of R-B
Verify ~5-10 ng of R-B in 6% TBE gel by running at 250 volts for 23 min with 0.5 µL of Low DNA Mass Ladder as a reference. CRITICAL STEP: If the smear of R-B is between 375 bp and above, continue to cDNA tagmentation. If there is strong smear smaller than 375 bp, repeat another round of 0.8x KAKA Pure Beads and elute with 40 µL of nuclease-free water.
Hash libraries: 2nd PCR, purification, and validation
2h
In PCR strip tubes, use 5 ng of R-A as template for enrichment of hashing libraries and adjust the volume of template to 10 µL with nuclease-free water and quick spin down the tubes. CRITICAL STEP: If the concentration of PCR product is higher than 5 ng/µL, it tends to have high error to pipette the volume smaller than 1 µL. We recommend to aliquot R-A, dilute into 0.5 ng/µL in total 30-50 µL and use 10 µL as template for 2nd PCR
Set up 2nd PCR mix during incubation
25 µL KAPA Hifi Hotstart ReadyMix, 2X
2.5 µL HASH_L0X, 10 uM
2.5 µL EvaGreen, 20X
7.5 µL Nuclease free water
Sequences of HASH_L0X (pick one of L01-3):
HASH_L01: AATGATACGGCGACCACCGAGATCTACACGTAACATGCGACACTCTTTCCCTACACGACG
HASH_L02: AATGATACGGCGACCACCGAGATCTACACGTGGATCAAAACACTCTTTCCCTACACGACG
HASH_L03: AATGATACGGCGACCACCGAGATCTACACCACTACGAAAACACTCTTTCCCTACACGACG
Add 37.5 µL of PCR mix and 2.5 µL of 10 µM SPLiT_N7XX (attached tables) primers into the tube with R-A template
2.5 µL SPLiT_N7XX, 10 uM
Run PCR on thermocycler using program follows: 72 ºC for 3 min, 98 ºC for 30 s, 9-12 cycles of (98 ºC for 30 s, 63 ºC for 30 s, 72 ºC for 45 s), 72 ºC for 3 min, 4 ºC hold. Terminate the reaction when it reaches mid-exponential growth.
Pool all reactions together, purify an aliquot of 50 uL with 1X beads. Elute purified hash libraries with 30 µL of nuclease-free water.
1. Binding
50 µL KAPA Pure Beads 00:08:00
2. Washing
180 µL Ethanol, 100% 00:00:30
3. 2nd wash
180 µL Ethanol, 100% 00:00:30
4. Drying
00:01:00 37 °C
5. Elution
30 µL Nuclease free water 00:10:00 37 °C
20m
Use 1 µL of hash sequencing libraries to determine for DNA concentration with Qubit dsDNA HS assay
Verify ~ 5 ng of hash libraries in 6% TBE gel at 250 volts for 23 min with 0.5 µL of Low DNA Mass Ladder as a reference.
sn/scRNA libraries: cDNA tagmentation and library preparation
3h
Mix ME and Nextera Adapter 1 (Ad1) oligos 1:1 for final 50 uM each. Briefly vortex and quick spin the tube. Anneal transposons on thermocycler using following program: 95 ºC 5 min, slowly cool down to 65 ºC (0.1 ºC/s), 65 ºC 5 min, slowly cool down to 4 ºC (0.1 ºC/s) and hold at 4 ºC.
Load Tn5 (In-house, 11.32 uM)
20 µL Tn5, (In-house, 11.32 uM)
6.8 µL 50 uM, Nextera Adapter 1/ME
Incubate 1 hour at room temperate with gentle shaking.
01:00:00
1h
Prepare Tagmentation mix as follows in 1.5 mL tube and aliquot 18 µL per pool into 0.2 mL PCR strip tubes with hinged cap.
2 µL Tango Buffer, 10X
2 µL DMF, 100%
12 µL Nuclease free water
2 µL Loaded Tn5, 8.44 uM
Transfer 2 µL of diluted cDNA (total 10-20 ng) with multichannel pipette into tagmentation mix, mix by pipetting 10 times, gently vortex and quick spin on mini centrifuge at room temperature for 5 s
Place the tubes on thermocycler that set at 55 ºC for 7 min with the heated lid
00:07:00
7m
Remove the tube from thermocycler and stop reaction by adding 5 µL of 0.2 % (wt/vol) SDS, mix by pipetting 5 times, gently spin down the tubes on mini centrifuge for 5 s and incubate at room temperature for 5 min
5 µL 0.2% SDS
00:05:00
Set up Tagmentation PCR mix during incubation
25 µL KAPA Hifi Hotstart ReadyMix, 2X
2.5 µL BC_0118, 10 uM
2.5 µL EvaGreen, 20X
7.5 µL Nuclease free water
Add 2.5 SPLiT_N7XX (attached tables) primers to 10 uL tagmented cDNA, then 37.5 of Tagmentation PCR mix respectively, mix well and gently spin down the tube on mini centrifuge for 3 s
2.5 µL SPLiT_N7XX, 10 uM
10 µL tagmented cDNA
37.5 µL Tagmentation PCR Mix
Run PCR on thermocycler using program follows: 72 ºC for 3 min, 98 ºC for 30 s, 9-12 cycles of (98 ºC for 30 s, 63 ºC for 30 s, 72 ºC for 45 s), 72 ºC for 3 min, 4 ºC hold. Terminate the reaction when it reaches mid-exponential growth.
20m
Purify PCR products at least two rounds of 0.7x KAPA Pure beads to make sure that no adapter dimers are leftover in the sequencing libraries. Elute the last round of bead purified RNA libraries with 30 µL of nuclease-free water. CRITICAL STEP: Adapter dimers can cluster more efficiently than regular sequencing libraries and is more sensitive on the patterned flow cell such as NovaSeq S4. This can lead to the reduction of sequencing output and sequencing quality
1. Binding
35 µL KAPA Pure Beads 00:08:00
2. Washing
180 µL Ethanol, 100% 00:00:30
3. 2nd wash
180 µL Ethanol, 100% 00:00:30
4. Drying
00:01:00 37 °C
5. Elution
50 µL Nuclease free water 00:10:00 37 °C
6. Binding
35 µL KAPA Pure Beads 00:08:00
7. Washing
180 µL Ethanol, 100% 00:00:30
8. 2nd wash
180 µL Ethanol, 100% 00:00:30
9. Drying
00:01:00 37 °C
10. Elution
30 µL Nuclease free water 00:10:00 37 °C
Use 1 µL of RNA sequencing libraries to determine for DNA concentration with Qubit dsDNA HS assay
Verify ~ 5 ng of RNA libraries in 6% TBE gel at 250 volts for 23 min with 0.5 µL of Low DNA Mass Ladder as a reference
sn/scRNA and hash libraries: Quality validation by MiSeq sequencing
1d
Validate sequencing libraries with MiSeq sequencing using v3 reagent kit by loading at 22 pm based on Qubit dsDNA HS quantification with at least 5% PhiX spike. The sequencing configuration is 70 cycles for read 1, 6 cycles for index, and 102 cycles for read 2.a
Note
CRITICAL STEP: For MiSeq sequencing, quantification of sequencing library concentration by Qubit and determination of average library size is sufficient. For high-throughput sequencing, we recommend to run sequencing libraries on Bioanalyzer, TapeStation or equivalent instrument that can determine accurate average library size and contamination of adapter dimers peaks at approximately 125-175 bp. We recommend running qPCR to quantify sequencing library concentration using the average library size derived from TapeStation. For a two-channel sequencing system such as NovaSeq, we recommend spike-in with a minimum of 10% PhiX or consult sequencing core.
Mix 1:1 ratio of "SN2-R R1" sequencing primer pairs for sequencing read 1 on Illumina workflow A (HiSeq 2500, MiSeq, NovaSeq 6000 platforms).
3 µL SNARE2_Read1, 100 uM
3 µL PhiX_Read1, 100 uM (Hash libraries uses the same Read 1 sequencing primer as PhiX)
594 µL Hybridization buffer, HT1
Mix 1:1 ratio of "SN2-R Index1" sequencing primer pairs for sequencing index 1 on Illumina workflow A (HiSeq 2500, MiSeq, NovaSeq 6000 platforms).
3 µL SNARE2-R_Index1
3 µL PhiX_Read1
594 µL Hybridization buffer, HT1
Mix 1:1 ratio of "SN2-R R2" sequencing primer pairs for sequencing read 2 on Illumina workflow A (HiSeq 2500, MiSeq, NovaSeq 6000 platforms).
3 µL SNARE2-R_Read2
3 µL PhiX_Read2
594 µL Hybridization buffer, HT1