Dec 02, 2024
Version 2
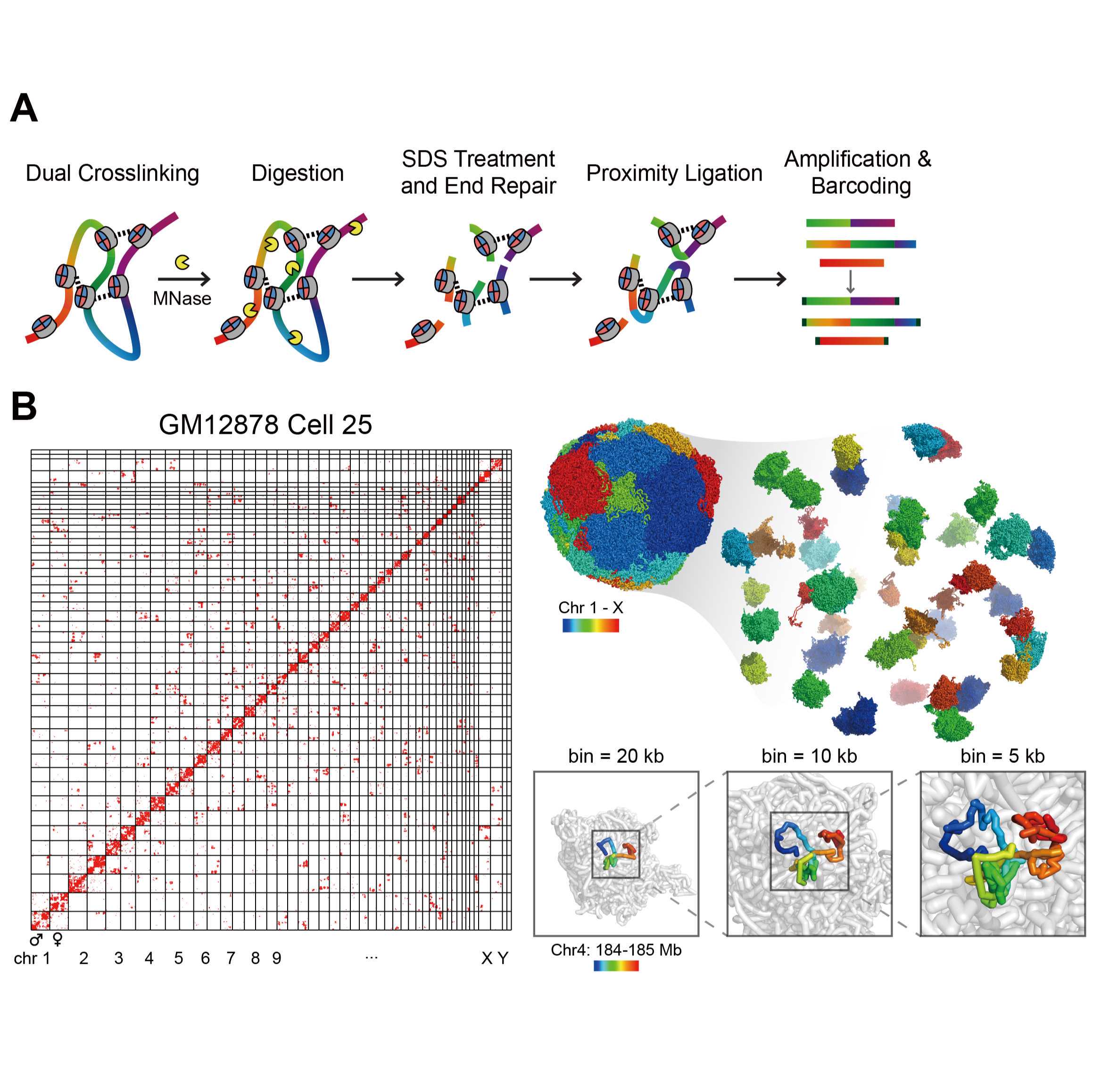
single-cell Micro-C protocol V.2
- 1Biomedical Pioneering Innovation Center, Peking University, Beijing 100871, China;
- 2Department of Neurobiology, Stanford University, Stanford, CA 94305, USA



Protocol Citation: Honggui Wu, Longzhi Tan 2024. single-cell Micro-C protocol. protocols.io https://dx.doi.org/10.17504/protocols.io.kqdg39wbzg25/v2Version created by Honggui Wu
Manuscript citation:
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: December 02, 2024
Last Modified: December 02, 2024
Protocol Integer ID: 113430
Keywords: single-cell 3D genome structure, single-cell Hi-C, chromosome conformation, Micro-C, cell 3d genome mapping assay, scale chromatin folding in individual cell, accurate detection chromatin loop, chromatin with an ionic detergent, scale chromatin folding, cell micro, genome amplification method, scale chromatin structure, help of micrococcal nuclease, micrococcal nuclease, chromatin loop, chromatin structure, reproducible reconstruction of 3d genome structure, 3d genome structure, genome, transcription factor, proper dna fragment, individual cell, cell, whole genome amplification
Abstract
Here we present a single-cell 3D genome mapping assay with improved spatial resolution with the help of micrococcal nuclease (MNase), termed single-cell Micro-C (scMicro-C). To achieve scMicro-C, we made three improvements. First, we titrated MNase digestion to reduce DNA loss and produce proper DNA fragments. Second, we solubilized chromatin with an ionic detergent, sodium dodecyl sulfate (SDS), which dramatically improved ligation efficiency. Third, we adopted our high-coverage transposon-based whole-genome amplification method, META (19), using Tn5 (20), to enhance the detection of chromatin “contacts.” scMicro-C retains the high signal-to-noise and high-resolution characteristics of bulk Micro-C, as indicated the accurate detection chromatin loops and other fine-scale chromatin structures. Furthermore, scMicro-C retains nucleosome and other chromatin-bound proteins (e.g., transcription factor) occupancy information. With our previously developed Dip-C algorithm, we confirmed that scMicro-C enables the reproducible reconstruction of 3D genome structures in single-cell at 5 kb resolution. With such kilobase 3D structures, we were able to explore the fine-scale chromatin folding in individual cells.
Crosslink Cells for Micro-C
Prepare 1 x BSA in PBS for Washing.
- Add 50 µL 10% BSA (MACS 130-091-376, final 1x) to 50 mL 1x PBS.
Prepare 1% Paraformaldehyde (1 mL for 1 million cells), recover to room temperature.
- 1 mL 32% PFA (EMS 15714, final 1%)
- 31 mL 1x PBS
Prepare 3 mM DSG (1 mL for 1 million cells).
- Dissolve 50 mg DSG (Thermo Fisher 20593) with 511 µL DMSO to a final concentration of 300 mM.
- Add 300 µL 300 mM DSG to 29.7 mL 1 x PBS to a final concentration of 3 mM, protected from light.
Harvest cells.
- Collect cell growth with appropriate density and high viability.
- Wash cells once with 1x PBS.
Fixation.
- Resuspend cell pellets in 1% PFA, and incubate at room temperature with rotation for 10 min.
- At the end of incubation, add 2 M Tris-HCl ph 7.5 to a final concentration of 0.75 M, and incubate at RT for 5 min.
- Centrifuge at 3000 x g for 5 min in a swing bucket centrifuge.
Remove the supernatant, then wash twice with 5 mL ice-cold 1 x BSA in PBS.
Crosslink with DSG.
- Resuspend in 3 mM DSG, and incubate at RT for 45 min with rotation.
- At the end of incubation, add 2 M Tris-HCl ph 7.5 to a final concentration of 0.75 M, and incubate at RT for 5 min.
- Centrifuge at 3000 x g for 5 min in a swing bucket centrifuge.
Count cells and aliquot.
- Remove the supernatant, then wash twice with 5 mL ice-cold 1 x BSA in PBS.
- Count cell number, then aliquot 1 million cells each to 1.5 mL tube.
- Store at -80°C (up to a year).
Prepare Micro-C Buffers
Prepare Micro-C Buffers
10 mL Prepare Micro-C Buffer 1 (300 µL per sample )
- 50 µL 5 M NaCl (Invitrogen AM9760G; final 50 mM)
- 50 µL 1 M Tris pH 7.5 (Invitrogen 15567027; final 10 mM)
- 25 µL 1 M MgCl2 (Invitrogen AM9530G; final 5 mM)
- 5 µL 1 M CaCl2 (Sigma 21115; final 1 mM)
- 50 µL 5% Digitonin (Sigma D141-100MG, final: 0.05%)
- 50 µL 100 x protease inhibitor (Roche 4693159001) (final: 1 X)
- 4.77 mL nuclease-free H2O
Prepare Micro-C Buffer 2 (10 mL )
- 100 µL 5 M NaCl (Invitrogen AM9760G; final 50 mM)
- 100 µL 1 M Tris pH 7.5 (Invitrogen 15567027; final 10 mM)
- 100 µL 1 M MgCl2 (Invitrogen AM9530G; final 10 mM)
- 9.7 mL nuclease-free H2O
Prepare Micro-C Buffer 3 (10 mL )
- 500 µL 1 M Tris pH 7.5 (Invitrogen 15567027; final 50 mM)
- 100 µL 1 M MgCl2 (Invitrogen AM9530G; final 10 mM)
- 9.4 mL nuclease-free H2O
MNase Digestion
Dilute MNase.
- Make MNase Dilution Buffer (10 mM Tris-HCl ph 7.5, 50 mM NaCl, 1 mM EDTA, 50% Glycerol).
- Aliquot 10,000 Unit MNase (NEB M0247S, 2,000 U/µL) to 500 µL MNase Dilution Buffer to a final concentration of 20 U/µL.
- Store at -80°C for several month.
Thaw 5 tubes of cell pellets on ice.
Resuspend in 100 µL Micro-C Buffer 1, incubate on ice for 20 min, and aspiration every 5 min.
Centrifuge at 800 x g at 4°C for 5 min. Carefully discard supernatant.
MNase Digestion
Then resuspend the pellet in 100 µL Micro-C Buffer 1, perform a titration of MNase to determine the optimal MNase concentration. For example, if start with 1 million cells, we will test 20U, 50U, 100U, 150U, 200U, 400U, add corresponding volume of MNase to each tube (a titration, typically for 1 million cells, the optimal MNase is 200U), then incubate at 37°C for 10 min.
At the end of incubation, add 2 µL 0.5 M EGTA to stop the reaction.
MNase Digestion
Take 5 ul to check digestion efficiency, remaining centrifuge at 800 x g at 4°C for 5 min. Carefully discard the supernatant, and wash once with 500 µL Micro-C Buffer 2. Store at -80°C (stable for up to several weeks) or proceed to End Repair.
Check Digestion Efficiency
- Take 5 µL above sample to check digestion efficiency.
- Add 85 ul 1 x PBS, 5 ul 10% SDS (Sigma, 71736-100ML), and 5 ul 20 mg/mL ProteaseK (QIAGEN, 19131).
- incubate at 65°C for 2 hr.
- Purify with DNA clean column (ZYMO, D4014).
- Run capillary electrophoresis (Fragment Analyzer/TepeStation/Bioanalyzer) to analyze digestion efficiency.
An exemplified MNase titration experiment: fragment size distribution with different MNase concentrations.
End Repair
Thaw the cell pellet (with the most appropriate fragment size) on ice.
Prepare 0.5% SDS.
- Add 25 µL 10% SDS to 475 µL Micro-C Buffer 2 to a final concentration of 0.5%.
Solubilize Chromatin.
- Resuspend the cell pellets in 50 µL 0.5% SDS.
- Incubate at 62°C for 10 min.
- At the end of incubation, add 50 µL 5% Triton X-100 to quench SDS. Incubate at 37°C for 15 min.
Centrifuge at 800 x g for 5 min, carefully remove supernatant.
Prepare End Repair Mix 1 (45 µL each sample).
- 5 µL 10 X NEBuffer 2.1 (NEB B7202S)
- 1 µL 100 mM ATP (Thermo R0441)
- 2.5 µL 100 mM DTT (final: ~5 mM)
- 2.5 µL 10 U/uL T4 PNK (NEB M0201S) (final: ~0.5 U/uL)
- 34 µL nuclease-free H2O
- Resuspend in 45 µL End Repair Mix1, and incubate at 37°C for 15 min.
- At the end of incubation, add 5 µL 5 U/uL Klenow Fragment (NEB M0210S), incubate at 37°C for another 15 min.
Prepare End Repair Mix2 (25 µL each sample)
- 2.5 µL 10 X T4 DNA Ligase Buffer (NEB B0202S)
- 0.5 µL 10 mM (each) dNTP (NEB N0447S)
- 0.125 µL 20 mg/mL BSA (NEB B9000S)
- 21.875 µL nuclease-free H2O
After Klenow incubation, add 25 µL End Repair Mix2 to each tube, incubate at 23°C for 45 min (shake 30 s and pause 2 min).
Add 5 ul 0.5 M EDTA (Invitrogen AM9260G).
Incubate at 65°C for 20 min.
Wash once with 1 mL Micro-C Buffer 3.
Proximity Ligation
Prepare Ligation Mix (250 µL each sample).
- 25 µL 10 X T4 DNA Ligase Buffer (NEB B0202S)
- 1.25 µL 20 mg/mL BSA (NEB B9000S)
- 12.5 µL 400 U/uL T4 DNA Ligase (NEB M0202S)
- 211.25 µL nuclease-free H2O
Resuspend the cell pellet in 250 µL Ligation Mix.
Take 25 µL as ligation efficiency control.
Check Ligation Efficiency
- Take 25 µL ligation product above sample to check ligation efficiency.
- Add 65 ul 1 x PBS, 5 ul 10% SDS (Sigma, 71736-100ML), and 5 ul 20 mg/mL ProteaseK (QIAGEN, 19131).
- incubate at 65°C for 2 hr.
- Purify with DNA clean column (ZYMO, D4014).
- Run capillary electrophoresis (Fragment Analyzer/TepeStation/Bioanalyzer) to analyze ligation efficiency.
A typical fragment size distribution after digestion and ligation.
Single-cell Amplification
Primers
- META 16 sequence
| A | B | |
| META-16-1 | GGCACCG AAAA | |
| META-16-2 | CTCGGCGA TAAA | |
| META-16-3 | GGTGGAGC ATAA | |
| META-16-4 | CGAGCGCA TTAA | |
| META-16-5 | AGCCCGGT TATA | |
| META-16-6 | TCGGCACC AATA | |
| META-16-7 | GCCTGTGG ATTA | |
| META-16-8 | GCGACCCT TTTA | |
| META-16-9 | GCATGCGG TAAT | |
| META-16-10 | GCGTTGCC ATAT | |
| META-16-11 | GGCCGCAT TTAT | |
| META-16-12 | ACCGCCTC TATT | |
| META-16-13 | CCGTGCCA AAAT | |
| META-16-14 | TCTCCGGG AATT | |
| META-16-15 | CCGCGCTT ATTT | |
| META-16-16 | CTGAGCTCG TTTT |
- META Transposon
| A | B | |
| META Transposon | 5'-[META sequence]-AGATGTGTATAAGAGACAG-3' | |
| 19 bp ME | 5'-/phos/-CTGTCTCTTATACACATCT-3' |
- First Step PCR Primers
| A | B | |
| First PCR primers | 5'-[META sequence]-AGATGTGTATAAG-3' |
Here we use 12 x 8 barcode combinations to distinguish individual cells.
- META16-ADP1 cell barcode
| A | B | |
| 1-1 | GATATG | |
| 1-2 | ATACG | |
| 1-3 | CCGTCTG | |
| 1-4 | TGCG | |
| 1-5 | GAACTCG | |
| 1-6 | ATGTAG | |
| 1-7 | CCCG | |
| 1-8 | TATGT | |
| 1-9 | GAGTAAG | |
| 1-10 | ATCG | |
| 1-11 | CCTAG | |
| 1-12 | TGACCG |
- META16-ADP2 cell barcode
| A | B | |
| 2-1 | ACTCTA | |
| 2-2 | AGAGCAT | |
| 2-3 | GGTATG | |
| 2-4 | TCGATGC | |
| 2-5 | CTACTAG | |
| 2-6 | TATGCA | |
| 2-7 | CACACGA | |
| 2-8 | GTCGAT |
Second Step Barcoded PCR Primers
| A | B | |
| META16-ADP1 | 5'-CTTTCCCTACACGACGCTCTTCCGATCT-[cell barcode]-[META sequence]-AGATGTGTATAAG-3' | |
| META16-ADP2 | 5'-GAGTTCAGACGTGTGCTCTTCCGATCT-[cell barcode]-[META sequence]-AGATGTGTATAAG-3' |
Prepare Reagents.
Prepare 60 mg/mL Protease.
- Dissolve 1 vial QIAGEN protease (QIAGEN 19155) in 2.78 mL 50% glycerol.
- Aliquot and store at -80°C (stable for up half a year).
Cell Triton Lysis Buffer (1 mL ).
- 10 µL 1 M Tris pH 8.0 (Invitrogen AM9855G, final 10 mM)
- 4 µL 5 M NaCl (Invitrogen AM9760G, final 20 mM)
- 2 µL 0.5 M EDTA (Invitrogen AM9260G, final 1 mM)
- 10 µL 10% Triton X-100 (Sigma 93443, final 0.1%)
- 25 µL 1 M DTT (Sigma 646563, final 25 mM)
- 5 µL 100 µM Carrier ssDNA (final 100 nM)
- Add H2O to 1 mL
- 25 µL 60 mg/mL QIAGEN protease (QIAGEN 19155, final 1.5 mg/mL) (freshly add when used)
| A | B | |
| Carrier ssDNA | TCAGGTTTTCCTGAA |
2 X Transposition Buffer (1 mL )
- 20 µL 1 M Tris-HCl pH 8.0 (Invitrogen AM9855G, final 20 mM)
- 10 µL 1 M MgCl2 (Thermo Fisher AM9530G, final 10 mM)
- 400 ul 40% (w/w) Polyethylene glycol 8000 (Sigma P1458, final 16% (w/w) )
- 570 µL nuclease-free H2O
Store at -20℃ if needed
Pick single cells.
Use mouth pipette or Fluorescence-activated cell sorting (FACS) to pick single cell into single PCR tubes or 96-well plates containing 2 µL cell lysis buffer.
Cell lysis.
Incubate as:
50℃, 1 hr
65℃, 1 hr
70℃, 15 min
Stored at -80℃ or proceed to transposon-based amplification.
Transposition (8 µL for each well, below recipe for 1 96-well plate).
- Prepare Transposition Mix
- 440 µL 2 X Transposition Buffer
- Add META transposome* to a final concentration of 0.3 nM
- Add nuclease-free H2O to 880 µL
* Transposome: META transposome is optional. Alternatively, Nextera transposome can be used (if using Nextera procedure, please refer to "Dip-C (Part 2: Whole-genome Amplification with Nextera)") on protocol.io.
2. Add 8 µL Transposition Mix to each well with a multi-channel pipette, vortex to mix.
3. Incubate at 55℃ for 10 min.
Tn5 Stop.
- Prepare Tn5 Removal Mix (250 mM NaCl, 37.5 mM EDTA, 2 mg/mL QIAGEN protease).
- Add 2 µL Tn5 Removal Mix to each tube, and mix thoroughly.
- Incubate as:
50℃, 30 min
70℃, 15 min
Pre-amplification.
Prepare Preamplification Mix (16 µL each well, below recipe for 1 96-well plate)
- 1540 µL 2 X Q5 High-fidelity Master Mix (NEB M0492L)
- 98.6 µL 50 µM First Step PCR Primers (final 0.1 µM each)
- 55 µL 100 mM MgCl2
- 66.4 µL nuclease-free H2O
Add 16 µL Preamplification Mix each well with a multi-channel pipette, and vortex to mix.
Amplified as:
72℃, 5 min
98℃, 30 s
12 cycles of [98℃ for 10 s, 62℃ for 30 s, 72℃, 2 min]
65℃, 5 min.
Cell Barcoding PCR.
- Add 1 µL individual 50 μM barcoded META16-ADP1 Primer and 1 µL individual 50 μM indexed META16-ADP2 Primer Mix to each well with a multi-channel pipette.
- Incubate as:
98℃, 30 s
3 cycles of [98℃ for 10 s, 62℃ for 30 s, 72℃, 2 min]
65℃, 5 min
Illustration of barcode addition.
Purification.
- Pool a 96-well plate to purify.
- Purify with clean column (ZYMO, D4014).
- Quantify with Qubit Fluorometer.
Library preparation.
- Take 400 ng amplified product for each plate as input.
- Prepare Library Preparation Mix (50 µL 2 X Q5 High-fidelity Mater Mix, template (400 ng), 5 µL 10 µM Indexed i5 primer, 5 µL 10 µM Indexed i7 primer (Vazyeme N321/N322), nuclease-free H2O to 100 µL )
- Amplified as:
98℃, 30 s
2 cycles of [98℃, 10 s, 68℃, 30 s, 72℃, 2 min]
72℃, 5 min
- Purify with PCR clean column (ZYMO D4014), then purify with 0.75 x AMPUre XP beads to select > 300 bp fragments for sequencing.
Sequencing.
META library only has 20% valid fragments having both P7 and P5 on either side, need to RUN qPCR quantify before loading to Illumina sequencing chip.