Jan 14, 2025
Version 4
SHARE-seq protocol v2.2 V.4
- Amelia Hall1,
- Gwyneth Torrecampo1,
- Alexandra Ham1,
- Cassandra White1,
- Guillermo Barreto Corona1,
- Eugenio Mattei1,
- Charles B Epstein1
- 1Broad Institute of MIT and Harvard
- IGVF



Protocol Citation: Amelia Hall, Gwyneth Torrecampo, Alexandra Ham, Cassandra White, Guillermo Barreto Corona, Eugenio Mattei, Charles B Epstein 2025. SHARE-seq protocol v2.2. protocols.io https://dx.doi.org/10.17504/protocols.io.81wgbx1oylpk/v4Version created by Amelia Hall
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: January 14, 2025
Last Modified: January 14, 2025
Protocol Integer ID: 118249
Keywords: multi-omic, RNA, ATAC, single cell, plate based hybridization, version of the protocol share, seq, gene regulation observatory, gene regulation observatory at the broad institute, protocol share, epigenomics platform, protocol, igvf project, gene, link to the original paper, share
Funders Acknowledgements:
NIH
Grant ID: HG011986
Disclaimer
Updates in progress, however this version is stable and reflects how we process cells/nuclei in the lab.
Abstract
An updated version of the protocol SHARE-seq, as used by the Epigenomics Platform and Gene Regulation Observatory at the Broad Institute in the service of data production for the IGVF project. Link to the original paper and protocol here: https://www.sciencedirect.com/science/article/pii/S0092867420312538
Materials
Before start
Some general notes and guidelines (many thanks to Liz Rebboah, Ryan Weber, and Ali Mortazavi for their thoughtful comments and suggestions). A slightly irreverent way to describe SHARE would be "punch a bunch of holes in a bag (the cell/nucleus), manipulate it a lot, hope it doesn't fall apart." That is to say: this is a lossy protocol! Whatever you start with, you'll probably only retain 10-25% of the starting material by the end of the first day. One of the biggest challenges of SHARE is keeping the RNA quality high (the ATAC is more stable). To support this, at the Broad we generally aliquot the following reagents: DTT, 10x SYBR, proteinase K, PIC(in 0.2mL PCR tubes), BSA (in 5ml tubes). We also aliquot buffer stocks (like Tris, MgCl2, NaCl) into 50mL or 15mL conical tubes. If there are any issues with the RNA capture or quality (as revealed by running gels on the second day of SHARE), throw out your current in use aliquot or stocks! Then you can balance a need for RNase free reagents with avoiding waste.
1.1 Ordering Oligo Plates and oligos for plate production
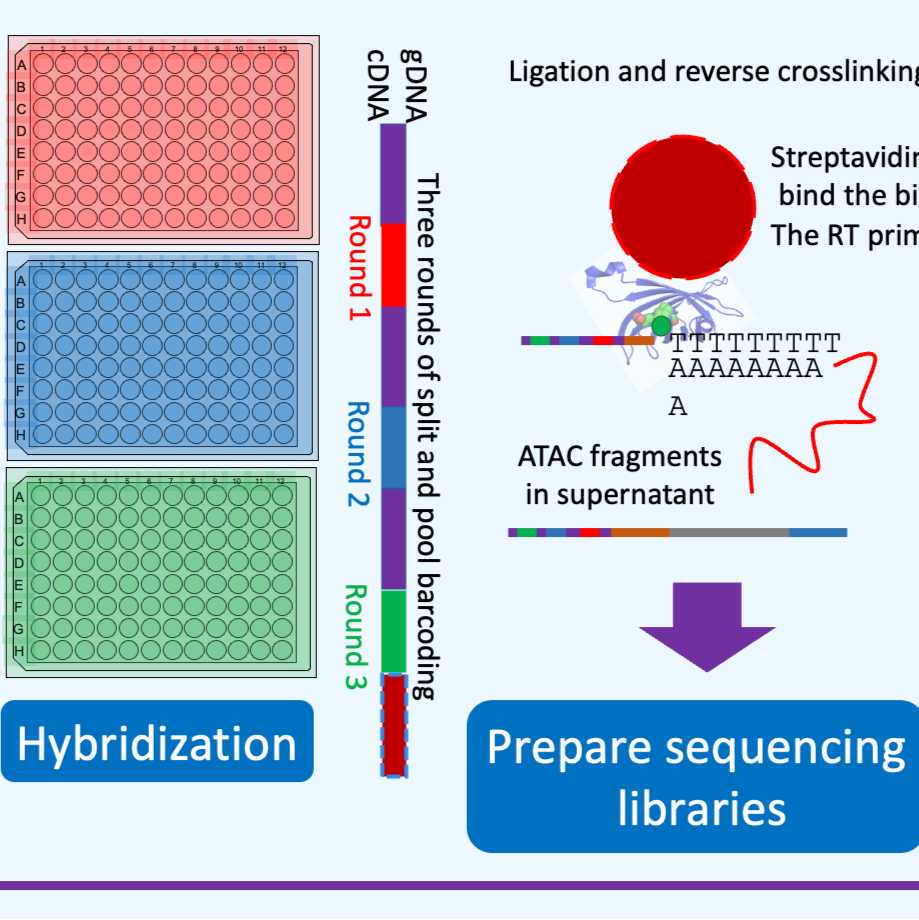
A visual overview of SHARE-seq:
This image shows a workflow for SHARE-seq
One of the more involved aspects of SHARE-seq (and SPLiT-seq) is properly making the oligo hybridization 96 well plates - this section covers that in detail before moving into any of the day to day aspects of this protocol (those start at step 24, in section 2).
In summary, this section will walk you through ordering deepwell 96 well oligo plates for all 3 rounds of hybridization in SHARE-seq (or SPLiT-seq), with an additional set of 96 oligos that can be ordered ("plate 2"), plus linker oligos.
In the original SPLiT-seq paper the authors calculated a barcode collision rate above ~5% with greater than around 25,000 cells or nuclei in a cellular sub pool (these are generated at the end of the first day of SHARE-seq). So for experiments using a singular set of three rounds of 96 barcodes, don't create cellular sub pools greater than 25,000 cells - the rate of barcode collisions will be high. If a second plate of 96 barcodes is used (we refer to these as "plate2"), and the two plates are pooled together and intermixed after each round of hybridization, we determined that up to 100,000 cells or nuclei can be pooled together safely, with low (<5%) risk of barcode collision.
Ordering oligo deepwell plates from IDT:
Using the attached Excel file, batch order the oligos on the tabs labeled "Plate1 R1 96", "Plate1 R2 96" "Plate1 R3 96" from IDT in a 96 well "deepwell" plate. If 192 barcodes are desired (2 plates), then order the oligos on the tabs labeled "Plate2 R1 96", "Plate2 R2 96", "Plate2 R3 96." Order at 25nM yield, 100uM concentration, resuspended in IDTE buffer, in a 96 well plate format. An example of what the plate upload looks like is shown below. Note that the R3 plate(s) will be less expensive than the R1 or R2 plates, since there is no 5' phosphate base needed in these molecules.
What it looks like to select plates from the above excel file for IDT batch plate ordering.
Ordering specifications for all plates ordered
You'll also need linkers to anneal with the oligos in each plate to create the unique cellular barcode during the hybridization of SHARE. Order the oligos below from IDT, order the linker oligos at 1mM (1000uM) concentration, and the blocking oligos at 100uM; order oligos resuspended in IDTE.
Note: the 1uM is the "scale" of what you're ordering from IDT, not the concentration! The concentration is "formulation" from IDT, and that's 100uM in IDTE for the blocking oligos, and 1000uM in IDTE for the linker oligos.
| A | B | C | D | E | F | G | |
| Name | LENGTH | Sequence | Scale | Purification | Specific oligos | order from IDT | |
| Round 1 linker | 30 | CCGAGCCCACGAGACTCGGACGATCATGGG | 1um | STD | Round 1 linker,CCGAGCCCACGAGACTCGGACGATCATGGG,1um,STD | ||
| Round 2 linker | 30 | CAAGTATGCAGCGCGCTCAAGCACGTGGAT | 1um | STD | Round 2 linker,CAAGTATGCAGCGCGCTCAAGCACGTGGAT,1um,STD | ||
| Round 3 linker | 30 | AGTCGTACGCCGATGCGAAACATCGGCCAC | 1um | STD | Round 3 linker,AGTCGTACGCCGATGCGAAACATCGGCCAC,1um,STD | ||
| Round 1 blocking | 30 | CCCATGATCGTCCGAGTCTCGTGGGCTCGG | 1um | STD | Round 1 blocking,CCCATGATCGTCCGAGTCTCGTGGGCTCGG,1um,STD | ||
| Round 2 blocking | 30 | ATCCACGTGCTTGAGCGCGCTGCATACTTG | 1um | STD | Round 2 blocking,ATCCACGTGCTTGAGCGCGCTGCATACTTG,1um,STD | ||
| Round 3 blocking | 30 | GTGGCCGATGTTTCGCATCGGCGTACGACT | 1um | STD | Round 3 blocking,GTGGCCGATGTTTCGCATCGGCGTACGACT,1um,STD |
Table of linkers and blockers for construction of the SHARE-seq hybridization plates
1.2 Annealing Oligo plates - making oligo plates
This part occurs only after you have all of the oligo deepwell plates and the Round 1,2,3 linker oligos from IDT. If you don't have these yet, don't proceed with platemaking!
The recipe below is for eighteen 96 well plates for each round of barcoding (54 total plates per set of 96 oligos, that's 108 if you're making both plate1 and plate2). We recommend that you use different color PCR plates for each round of each plate. Conveniently, ThermoFisher offers 96 well skirted plates in 6 different colors, search for part number AB2396N (where N=R is red, G=green, Y=yellow, B=blue, O=orange - the base model is clear). Please be sure you purchase part number AB2396, and not AB3396 - those plates have white wells that will make it hard to tell how much fluid is in the well.
Thaw deepwell plates at RT for 20-40 minutes at RT, and spin down before usage to avoid oligo cross-contamination. Can speed the thaw by setting on a thermomixer set to 28ºC if needed.
Make 50mL of STE, recipe below:
| A | B | C | |
| Oligo Annealing buffer (STE) | Volume (mL) | 1x Concentration | |
| 1M Tris pH 8.0 | 0.5 | 10 mM | |
| 5M NaCl | 0.5 | 50 mM | |
| 0.5 M EDTA | 0.1 | 1 mM | |
| H2O | 48.9 | ||
| Total | 50 |
recipe for STE
Using a 12 channel multipipettor:
Dilute 180μl Round 1 linker oligo (1 mM, 1000uM) with 7920 μl STE buffer. Mix 80 μl diluted Round 1 linker oligo with 20 μl Round 1 oligo (100 μM) in a 96 well PCR plate.
Dilute 259.2 μl Round 3 linker oligo (1 mM, 1000uM) with 7047 μl STE buffer. Mix 72 μl diluted Round 3 linker oligo with 28 μl Round 3 oligo (100 μM) in a 96 well PCR plate.
Dilute 216μl Round 2 linker oligo (1 mM, 1000uM) with 7371 μl STE buffer. Mix 76 μl diluted Round 2 linker oligo with 24 μl Round 2 oligo (100 μM) in a 96 well PCR plate.
Seal the plates - we use an BioRad PX1 plate sealer, seal at 180 degrees C for 5 seconds. We use the specific foil seals for that sealer as well (part number 1814045). If you don't have a heat sealer, we recommend BioRad Microseal F foil seals (part number MSF1001), which we use for sealing the final oligo plates as well.
Anneal Round 1, Round 2, and Round 3 plates using the PCR cycling conditions below. The slow ramp is critical for this step!! The plates won't work without the exact cycling conditions below!
| A | B | |
| 95ºC | 2 min | |
| Slow ramp, -1ºC/cycle, 1 min per cycle | ||
| 20ºC | 2 min | |
| 4ºC | Forever | |
| Total | ~1h 26 min |
PCR cycling conditions for the oligo plate annealing
After annealing, check if there is significant water evaporation for the wells at the corners/sides of the wells. With the PX1 heat sealing, we don't find that this occurs, but if you used more standard foil seals, there may be some evaporation on the sides.
Add 100μl STE to each well of the annealed plate, mix 7-8x and transfer 100μl to another 96 well plate. These two plates are your "stock plates" from which you will aliquot into the multi-colored plates for each round of SHARE.
Aliquot 10ul from your stock plates (you'll have either 3 or 6 sets) to make an oligo plate. We use the Agilent Bravo to make this go faster, but it's fairly quick with a 12 channel pipette as well. Don't forget to: label the plates! The rounds have to go in order for hybridization to work! Also don't forget to put each "round" of oligo in a separate colored 96 well plate to reduce errors when selecting plates for the experiment.
Seal plates with foil and label using labels or marker (printed labels are faster if you've got them). Store the plates in rack at -30ºC, they are good for at least 6 months, and probably 12 months.
1.3 Ordering and annealing oligos for cDNA tagmentation
SHARE-seq generates long cDNA (presuming no nuclease contamination or other harms befall the RNA). In this state it is overlong to sequence, so we must tagment it using Tn5 complexed with Illumina Read 1 only. To do this, we use commercial Tn5 from Diagenode, and incubate it with annealed oligos that include Illumina Read 1, and block the ME_Comp region of that sequence.
Order the following oligos from IDT resuspended at 100uM in IDTE
| A | B | C | D | E | F | G | |
| Name | LENGTH | Sequence | Scale | Purification | Specific oligos | order from IDT | |
| Read1 | 33 | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG | 1um | HPLC | * | Read1,TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG,1um,HPLC | |
| Blocked_ME_Comp | /5Phos/C*T*G* T*C*T* C*T*T* A*T*A* C*A*/3ddC/ | 1um | HPLC | * | Blocked_ME_Comp,/5Phos/C*T*G* T*C*T* C*T*T* A*T*A* C*A*/3ddC/ ,1um,HPLC |
Read1 and Blocked_ME_Comp to order
Wait for them to arrive before proceeding!
Prepare tagmentation adapter mix in PCR tubes.
| A | B | |
| Tagmentation Adapter | ul | |
| 100uM Read1 | 26 | |
| 100uM Blocked_ME_Comp | 26 | |
| 1M Tris pH 8.0 | 0.52 | |
| 5M NaCl | 0.52 | |
| Total | 53 |
tagmentation adapter mix, ideally make several aliquots, the above recipe is for a single aliquot.
Anneal oligos in a thermal cycle as follows.
| A | B | |
| 85ºC | 2 min | |
| Slow ramp, -1ºC/cycle, 1 min per cycle | ||
| 20ºC | 2 min | |
| 4ºC | Forever | |
| Total | ~1h 14 min |
PCR cycling conditions for annealing the tagmentation adapter oligos
Heat a 150ul aliquot of RNase free 100% glycerol to 65ºC in a heat block or thermomixer (RNase zap the surfaces). Make two 50ul aliquots and equilibrate to RT. Mix the annealed tagmentation adapter with an aliquot of glycerol, respectively (i.e. mix each 50ul aliquot of adapter with a separate 50ul aliquot of glycerol). The annealed adapters can be immediately used.
1.4 Essential SHARE-seq buffers (make the day before performing an experiment)
Make each of the following buffers - note that for large experiments you may need more than one aliquot of these buffers.
| A | B | C | |
| Dilution buffer | Volume (ul) | Final Concentration | |
| 100% glycerol (wide-bore tips) | 500 | 50% | |
| 1M Tris pH 7.5 | 50 | 50 mM | |
| 5M NaCl | 20 | 100 mM | |
| 5mM EDTA | 20 | 0.1 mM | |
| 1M DTT | 1 | 1 mM | |
| 10% NP-40 | 10 | 0.1% | |
| H2O | 390 | ||
| Total | 1000 |
Dilution buffer recipe. This buffer is used to dilute Tn5 enzyme for transposition and tagmentation
| A | B | C | |
| 5x SMART RT buffer (adapted from smart-seq-3) | Volume (ul) | Final Concentration (of 5x) | |
| 1000mM (1M) DTT | 40 | 40 mM | |
| 1M Tris pH 8.3 | 125 | 125 mM | |
| 100 mM GTP | 50 | 5 mM | |
| 5M NaCl | 30 | 150 mM | |
| 1M MgCl2 | 12.5 | 12.5 mM | |
| H2O | 742.5 | ||
| Total | 1000 |
SMART reverse transcription buffer - adapted from the smart-seq-v3 protocol.
You can also make the NI (see step 28) the day before SHARE. We recommend making a fresh 50mL aliquot for every experiment as a safeguard against RNase contamination.
1.5 Ordering SHARE-seq oligos
All of the below oligos are necessary for performing SHARE-seq (mainly amplifying libraries on Day 2 of SHARE). Wait until these arrive to start the SHARE-seq protocol. All should be ordered formulated at 100uM in IDTE.
| A | B | C | D | E | F | G | |
| Name | LENGTH | Sequence | Scale | Purification | Specific oligos | order from IDT | |
| RT_primer | 61 | /5Phos/GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGNNNNNNNNNN/iBiodT/TTTTTTTTTTTTTTVN | 1um | RNASE | * | RT_primer,/5Phos/GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGNNNNNNNNNN/iBiodT/TTTTTTTTTTTTTTVN,1um,RNASE | |
| P7 | 24 | CAAGCAGAAGACGGCATACGAGAT | 100nm | STD | * | P7,CAAGCAGAAGACGGCATACGAGAT,100nm,STD | |
| RNA_PCR_primer | 23 | AAGCAGTGGTATCAACGCAGAGT | 100nm | STD | * | RNA_PCR_primer,AAGCAGTGGTATCAACGCAGAGT,100nm,STD |
DNA oligos for SHARE to order from IDT
When the RT primer arrives, aliquot into single use amounts (ex: for 1e6 cells, you would need ~300ul of the RT primer). This oligo is long and somewhat unstable, so minimize freeze thaw cycles.
| A | B | C | D | E | F | G | |
| Name | LENGTH | Sequence | Scale | Purification | Specific oligos | order from IDT | |
| TSO | 30 | AAGCAGTGGTATCAACGCAGAGTGAATrGrG+G | 1umR | RNASE | * | TSO,AAGCAGTGGTATCAACGCAGAGTGAATrGrG+G,1umR,RNASE |
TSO (Template Switching Oligo) RNA oligo for SHARE-seq
2.0 Washing and Counting Fixed cells or nuclei (time: 30-60m)
This section presumes cells or isolated nuclei have been fixed in formaldehyde (0.2% for cultured cells/nuclei, 1% for primary PBMCs) and have been frozen at -80 degrees as a dry pellet. For more details on nuclei isolation, see the attached files (will be made into protocols.io links as time permits):
Important note: if your samples are PBMCs or HSCs, you'll want to use the optional polyAdenylation module (section 4) and perform centrifugation at 1000xg.
BEFORE YOU BEGIN!!! Spray down your bench with RNase zap! Spray your pipettors with RNase Zap! Put on a surgical mask! Spray your gloves with RNase Zap all the time! I know this seems annoying, but RNA will degrade easily in this protocol, and taking precautions early will really help!
Put swinging bucket centrifuge rotor (Eppendorf # S-24-11-AT) in the refrigerable benchtop Eppendorf centrifuge (model #5430R) and cool down to 4ºC.
Prepare RNase free, sterile NIB-2RI, NI-2RI, NID-2RI (as well as NIB and NI). NIB and NI are stable for at least 6 months at room temperature. We always make a fresh aliquot of NI when making the 5x SMART RT buffer and dilution buffer.
| A | B | C | |
| Nuclei Isolation Buffer (NIB) | Volume (mL) | Final Concentration | |
| 1M Tris-HCl pH 7.5 | 0.5 | 10mM | |
| 5M NaCl | 0.1 | 10mM | |
| 1M MgCl2 | 0.15 | 3mM | |
| 10% NP40 | 0.5 | 0.1% | |
| H2O | 48.75 | ||
| Total | 50 |
NIB buffer recipe
| Nuclei Isolation (NI) | Volume (mL) | Final Concentration | |
| 1M Tris-HCl pH 7.5 | 0.5 | 10mM | |
| 5M NaCl | 0.1 | 10mM | |
| 1M MgCl2 | 0.15 | 3mM | |
| H2O | 49.25 | ||
| Total | 50 |
NI buffer recipe
| A | B | |
| NIB-2RI | Volume (ml) | |
| NIB | 1000 | |
| Enzymatic RI | 7 | |
| SUPERase RI | 7 |
NIB-2RI recipe. Around 100uL per sample is used for cells/nuclei
| A | B | C | |
| NI-2RI | Volume (ml) | Volume (ml) | |
| NI (per expt) | 1000 | 50000 | |
| Enzymatic RI | 6.25 | 125 | |
| SUPERase RI | 6.25 | 125 | |
| 7.5% BSA | 5.33 | 266.67 |
NI-2RI recipe - around 7mL per sample is used for cells/nuclei
Transfer fixed cells to a new 1.5mL tube coated with 7.5% BSA. If starting from a fixed pellet, use 500ul NI-2RI to resuspend and transfer to a BSA coated tube.
Preparing 1.5ml BSA coated tubes: Prepare a 1.5mL Eppendorf tube by adding 150ml 7.5% BSA, vortex, remove supernatant, let tube sit on ice briefly, remove any collected BSA with
a P20.
Spin 750xg, 4ºC, 5 minutes. Gently remove supernatant.
Wash with 500ul NID-2RI without disturbing pellet.
Spin 750xg, 4ºC, 5 minutes. Gently remove supernatant.
Resuspend in 50ul NID-2RI.
Add 100ul NIB-2RI to each sample, incubate for 5 minutes on ice to permeabilize the nuclei.
Add 900ul NI-2RI (NI-RI: 1200ul NI + 3ul Enzymatic RI) to each sample, mix by inverting
Spin 750xg, 4ºC, 5 minutes. Gently remove supernatant, using a P20 to remove as much NIB-2RI as possible.
Turn on a thermomixer (Eppendorf ThermoMixer C) for the ATAC Tn5 transposition, set to 37 degrees and put the 1.5mL block on the thermomixer (you should have a 96 well block for the hybridization step).
Resuspend in 50ul NI-RI (NI-RI: 1200ul NI + 3ul Enzymatic RI). SUPERase RI inhibits the Tn5 transposition, so using RI instead of 2RI is very important here.
Use 2.5ul cells and 7.5ul HCB-RI plus 10ul Trypan blue to count nuclei using the BioRad TC20 (part number 1450102) or a manual hemocytometer (we like the Reichert Bright-Line: http://hausserscientific.com/products/reichert_bright_line.html). Determine cell counts per mL and per ul, so the volume needed to get to 10000 nuclei is known. Multiply the count by 4, since the cells/nuclei are diluted 1:4 before adding trypan blue. The TC20 takes trypan blue dilutions into account, so divide the count per mL by 1000 to get the count per ul.
Determine the volume of cell suspension that is 10,000 cells (one ATAC transposition), and the total number of ATAC reactions (10,000 cells/reaction). We typically do SHARE using 25 ATAC reactions (250K cells) as the basic "unit" of SHARE.
3.0 ATAC Tn5 transposition (time: ~45m)
Assemble transposome by mixing assembled Tn5, and dilution buffer. Here we use volumes of ASSEMBLED Tn5 from the SeqWell produced Tn5 (Tagify: https://seqwell.com/tagify-umi-reagents/)
We use 0.6ul per reaction (10000 cells) (enzyme concentration is 10mM).
| A | B | C | |
| Transposome | Volume (ml) | Volume N=25 | |
| 1x SeqWell Tn5 (volume as of 01/27/2024) | 0.6 ✕ N reactions | 15ul | |
| Dilution buffer | 1.875 ✕ N reactions | 46.9ul | |
| Total | 2.5 ✕ N reactions | 61.9ul |
Recipe for assembling the Tn5 for tubes of ATAC reactions (25 reactions per 1.5mL tube max)
For every 250K of cells, prepare one of the above tubes. You could also prepare a master mix using 10% overage as well, but we do a single tube for every ATAC reaction tube to use a bit less of the Tagify reagent.
Calculate the total number of ATAC reactions for this experiment. Prepare 1x TB accordingly to the recipe below. Note that 1x TB buffer must be made fresh.
| A | B | C | |
| 1x TB | Volume (ml) ✕ samples (N) | Volume N=27.5 (per tube plus overage) | |
| 0.2M Tris-acetate | 8.25 ✕ N | 226.875 | |
| 5M K-acetate | 0.66 ✕ N | 18.15 | |
| 1M Mg-acetate | 0.5 ✕ N | 13.75 | |
| 100% DMF | 8 ✕ N | 220 | |
| H2O | 24.04 ✕ N | 661.1 | |
| PIC | 0.2 ✕ N | 5.5 | |
| Enzymatic RI | 0.85 ✕ N | 23.375 | |
| Total | 42.5 ✕ N | 1168.75 |
Recipe for 1xTB buffer
The total volume of cells for the N number of ATAC reactions should fit into a 50ul reaction. Determine the total volume of cells needed for the ATAC reactions, and then use NI to bring the total volume up to 50ul. Example: with 25 ATAC reactions (250K cells), and 13,920 cells per ul, use 250K/13920 to get 17.95ul of cells needed. Add 50ul-14.4ul = 32.05ul NI-RI to get 50ul total volume of cells.
Add 42.5 ✕ N ul of 1xTB to cells + NI (in 50ul total volume): 1062.5ul for 25 rxns/sample, incubate at RT for 10 minutes (this is a permeabilization step).
Add 2.5 ✕ N ul (61.9ul for 25 reactions) of assembled Tn5 to sample, mix well by pipetting.
In the 1.5mL tube, seal, shake in a thermomixer at 500rpm for 30 minutes, 37ºC.
Every 5 minutes, take the entire block off the thermomixer and invert to mix
This resets the timer on the thermomixer! So use an external timer
Add 500ul NID-2RI to each tube, spin down at 750xg, RT, 5 minutes.
Remove supernatant, wash with 500ul NI-2RI, spin down at 750xg, RT, 5 minutes.
Remove supernatant, resuspend cells in 60ul NI-2RI (from a single ATAC reaction tube).
Set thermomixer(s) to 23ºC for the hybridization reaction (after the RT step) and put the 96 well blocks on.
4.0 Optional PolyAdenylation module (time: ~45m)
If your samples are from hematopoietic compartment (PBMCs, HSCs), you’ll want to use the optional exogenous polyAdenylation module in this section before Reverse Transcription to improve RNA capture (since PBMCs have a lot of nucleases). If not, skip ahead to section 5.0 Reverse Transcription (starting at step 57).
For each sample, mix 60ul cells/nuclei in NI-2RI from the previous step with 240ul PolyA master mix (recipe below), and incubate at 37ºC for 15 minutes in a thermomixer without mixing. (each 1.5mL ATAC tube is a single polyA reaction).
| A | B | C | |
| polyA mix | Volume (ml) ✕ samples (N) | Volume per sample (60ul NI-2RI + nuclei) | |
| H2O | 21.1xN | 126.6 | |
| 5x SMART RT buffer | 10xN | 60 | |
| Enzymatic RI | 0.3xN | 1.8 | |
| SUPERase RI | 0.6xN | 3.6 | |
| rATPs (NEB:P0756S) | 5xN | 30 | |
| E coli PolyA (NEB: M0276S) | 3xN | 18 | |
| Cells/NI-2RI | 10xN | 60 | |
| Total | 50xN | 300 |
Polyadenylation master mix recipe
Add 200ul NID-2RI, spin down at 1000xg, 5 minutes, RT.
Remove supernatant and add 200ul NI-2RI, mix by inverting.
Spin down at 1000xg, 5 minutes, RT, remove as much supernatant as possible.
Resuspend so the total volume is 60ul NI-2RI.
Proceed to step 57 of the protocol below and continue with the RT reaction. Each polyA tube becomes a single RT reaction (7 0.2mL PCR tubes).
5.0 Reverse Transcription (time: 50m-1h)
Make the following master mix for reverse transcription:
| A | B | |
| Reverse transcription (RT) mix | Volume/ATAC tube (ul) | |
| 5x SMART RT buffer | 70 | |
| Enzymatic RI | 2.19 | |
| SUPERase RI | 4.38 | |
| dNTPs | 17.5 | |
| 100uM RT primer | 35 | |
| H2O | 10.94 | |
| 50% PEG | 105 | |
| Maxima H Minus Reverse Transcriptase (add right before RT reaction) | 35 | |
| Total | 280 |
recipe for the RT mix.
For each ATAC 25 reaction tube, add 280ul RT mix to 60ul cells/NI-2RI, aliquot 50ul to 7 wells in a 96 well plate. Then, run the RT thermocycler protocol as described below.
| A | B | C | |
| 50ºC | 10 min | hold to start | |
| 8ºC | 12 s | 3 cycles | |
| 15ºC | 45 s | cycle | |
| 20ºC | 45 s | cycle | |
| 30ºC | 30 s | cycle | |
| 42ºC | 2 min | cycle | |
| 50ºC | 3 min | cycle | |
| 50ºC | 5 min | hold after cycles | |
| Total | ~41 min |
Cycling conditions for the reverse transcription reaction: set the lid temp to 60ºC
Pool all reactions for a given sample, add 300ul NID-2RI per 7 PCR tubes, spin down at 750xg, 5minutes, RT in a 1.5mL Eppendorf tube coated with 7.5% BSA. Note: the pellet may look larger than before - this is due to BSA dropping out of solution.
Wash with 500ul NI-2RI, spin down at 750xg, 5minutes, RT
6.0 Hybridization
Remove supernatant and resuspend each sample in NI-2RI. The amount each sample is resuspended in depends on the number of samples, and plates, per experiment. Examples: for a single sample in a single plate (96 well): resuspend in 1152ul NI-2RI. For four samples on a single plate, resuspend each sample in 288ul NI-2RI.
| fraction of plate | ul NI-2RI | |
| 2 plates | 2304 | |
| 1 plate | 1152 | |
| 1/2 plate | 576 | |
| 1/3 plate | 384 | |
| 1/4 plate | 288 | |
| 1/8 plate | 144 |
NI-2RI volumes per sample by experiment size
Note: it is very important to thaw oligo plates to RT (~23ºC) before hybridization. Spin down plates before using. Coat all reservoirs used for pooling with 7.5% BSA.
Make the hybridization buffer:
| A | B | C | D | |
| Hybridization buffer | Volume (ml) | Stock concentration | Final Concentration | |
| H2O | 5510 | -- | -- | |
| T4 ligase buffer (10x) | 1152 | 10x | 1x | |
| Enzymatic RI | 28.8 | 20 U/ml | 0.05 U/ml | |
| SUPERase RI | 92.16 | 40 U/ml | 0.32 U/ml | |
| 7.5% BSA | 184.32 | 7.50% | 0.10% | |
| Total | 6967.28 | � |
Hybridization buffer recipe for two plates
| A | B | C | D | |
| Hybridization buffer | Volume (ml) | Stock concentration | Final Concentration | |
| H2O | 2755 | -- | -- | |
| T4 ligase buffer (10x) | 576 | 10x | 1x | |
| Enzymatic RI | 14.4 | 20 U/ml | 0.05 U/ml | |
| SUPERase RI | 46.08 | 40 U/ml | 0.32 U/ml | |
| 7.5% BSA | 92.16 | 7.50% | 0.10% | |
| Total | 3483.64 |
Hybridization buffer recipe for a single plate experiment
If you have a single sample: transfer the cells in NI-2RI into a BSA-coated reservoir suitable for an 8 or 12 channel multi-channel pipette. Add all of the hybridization buffer to the reservoir (adjust volume for 1 or 2 plates).
If you have multiple samples, put each sample in a separate BSA-coated reservoir, suitable for an 8-channel pipette (this makes loading samples across the 96 well plate easier). Add the hybridization buffer to each sample according to the table below:
| fraction of plate | ul Hybridization buffer | |
| 2 plates | 6967.28 | |
| 1 plate | 3483.64 | |
| ½ plate | 1741.82 | |
| ⅓ plate | 1161.21 | |
| ¼ plate | 870.91 | |
| ⅛ plate | 435.455 |
We find that using a 96 well plate map helps a lot for sample mapping! An example image is below, along with an open-access svg file of a 96 well plate.
An example of a plate to sample mapping file for the first round of hybridization. The five digit identifiers are lab-internal identifiers for a given aliquot of biological material.
For those who have access to Photoshop or Gimp, the *.psd files are editable templates. We recommend a making a layer to color the wells by sample identity.
Transfer sample(s) to Round 1 plate(s) as follows for each row of wells:
a. Set a 12-channel pipette to 45ul, mix 2-3x
b. Transfer 45ul of mixture to Round 1 plates and mix by pipetting gently 5-7x.
c. Change tips between columns loaded!
Seal and shake at 300rpm for 25 minutes at RT (23 deg C).
Prepare Blocking oligo 1 as below and transfer to reservoir.
| A | B | C | |
| Blocking oligo 1 | Volume (ul) | Volume (ul) | |
| Round_1_blocking (100uM) | 266.07 | 532.14 | |
| T4 ligase buffer (10x) | 221.76 | 443.52 | |
| H2O | 721.77 | 1443.54 | |
| Total | 1209.6 | 2419.2 |
Column B is for a single plate, and column C is for two plates.
Add blocking oligo mix as follows:
a. Using a 12-channel pipette, transfer 10ul Blocking oligo 1 to each well, pipetting up and down to mix thoroughly, changing tips between rows.
b. Seal and shake at 300rpm for 15 minutes at RT
Pool samples as follows:
a. Set a 12-channel pipette to 70ul, pipette each row up and down 2-3 times, pipette up and down 2-3 times as ejecting each row into a BSA-coated reservoir (avoid bubbles).
b. After all rows have been pooled, pipette up and down 4-5 times to mix the reservoir
Open Round 2 oligo plates, and transfer 55ul of pooled mixture to each well, pipetting to mix each row. Shake at 300rpm for 25 minutes at RT.
Prepare Blocking oligo 2 as below and transfer to reservoir.
| A | B | C | |
| Blocking oligo 2 | Volume (ul) | Volume (ul) | |
| Round_2_blocking (100uM) | 319.305 | 638.61 | |
| T4 ligase buffer (10x) | 221.76 | 443.52 | |
| H2O | 668.535 | 1336.86 | |
| Total | 1209.6 | 2419.2 |
Column B is for a single plate, and column C is for two plates.
Add blocking oligo mix as follows:
a. Using a 12-channel pipette, transfer 10ul Blocking oligo 2 to each well, pipetting up and down to mix thoroughly, changing tips between rows.
b. Seal and shake at 300rpm for 15 minutes at RT
Pool samples as follows:
a. Set a 12-channel pipette to 80ul, pipette each row up and down 2-3 times, pipette up and down 2-3 times as ejecting each row into a BSA-coated reservoir (avoid bubbles).
b. After all rows have been pooled, pipette up and down 4-5 times to mix the reservoir
Open Round 3 oligo plate, and transfer 65ul of pooled mixture to each well, pipetting to mix each row. Shake at 300rpm for 25 minutes at RT.
Prepare Blocking oligo 3 as below and transfer to reservoir.
| A | B | C | |
| Blocking oligo 3 | Volume (ul) | Volume (ul) | |
| Round_3_blocking (100uM) | 278.25 | 556.5 | |
| H2O | 931.35 | 1862.7 | |
| Total | 1209.6 | 2419.2 |
Column B is for a single plate, and column C is for two plates.
Add blocking oligo mix as follows:
a. Using a 12-channel pipette, transfer 10ul Blocking oligo 3 to each well, pipetting up and down to mix thoroughly, changing tips between rows.
b. Seal and shake at 300rpm for 15 minutes at RT.
7.0 Ligation (time: ~45m)
Using either a P1000, or a 12-channel pipette and a BSA coated reservoir, aspirate the contents of each well and pool into a 15mL conical tube coated with 7.5% BSA.
a. Total volume should be ~14mL (for a two plate experiment), but will fit in a single tube.
b. For a single plate experiment, the total volume would be 7mL
Spin down at 750xg for 5 minutes at RT in a swinging bucket rotor.
Discard supernatant and add 1mL NID-2RI.
Spin down at 750xg for 5 minutes at RT in a swinging bucket rotor.
Discard supernatant and add 1mL NI-2RI, without disturbing pellet (let flow down side of tube slowly)
Spin down at 750xg for 5 minutes at RT in a swinging bucket rotor.
Discard supernatnant and resuspend in 80ul NI-2RI. It's ok if there is a little bit of extra volume here - you'll just resuspend all of the cells into the ligation mix.
Mix sample with 320ul ligation mix (recipe below) and aliquot 50ul to 8 PCR tubes.
| A | B | |
| Ligation mix | Volume (ul) | |
| H2O | 253.7 | |
| Enzymatic RI | 3.2 | |
| SUPERase RI | 1 | |
| T4 ligase buffer (10x) | 40 | |
| T4 ligase 400U /ul | 20 | |
| 7.5% BSA | 2.13 | |
| Total | 320 |
Ligation mix recipe. Note that if you have multiple single plate experiments, you'll need one ligation mix per experiment. If you're doing a double plate experiment (192 barcodes), you just need the single ligation mix recipe.
Shake at 300rpm for 30 min at RT.
Pool samples in a 1.5mL Eppendorf tube coated with 7.5% BSA, spin down 750xg, 5 minutes, RT, discard supernatant.
Gently resuspend pellet with 1mL NI-2RI
Spin at 750xg, 5 minutes, RT.
Presume 80% loss of nuclei over the course of the protocol, so for every 20,000 retained nuclei, resuspend in 10ul (goal is ~2000 cells or nuclei/ul).
For example: With 1 million starting cells, presume 100K are retained, and resuspend in 50ul NI-2RI.
Using 2ul cells and 8ul NI-2RI, mix with 10ul trypan blue. Count intact nuclei in a manual hemocytometer.
To get a count per mL, multiply hemocytometer counts by 100K
(dilution factors: 2x = trypan blue, 5x = cell:NI dilution, 10,000x = hemocytometer dilution).
To get a count per ul, multiply counts by 100 (conversion: 100 = 100,000 (dilution factor for hemocytometer plus dilutions)/1,000 (microliters in one mL)).
8.0 Reverse crosslinking-pull down (time: 1h15m)
Here you will aliquot cells into cellular or nuclear "sub pools", and then digest the remaining proteins away to leave only barcoded nucleic acids. The number of cells per pool strongly depends on the experimental setup: a 96 barcode (single plate) experiment should not contain more than 25,000 cells or nuclei - the risk of barcode collisions (two different cells with the same randomly generated barcode) increases substantially (>2.5%). The math here is 96*96*96=884,000, and 884K*0.025 = 22,118.
A 192 barcode (two plate) experiment has a maximum cell count of 100,000 cells per cellular or nuclear sub pool, though theoretically 200,000 should be possible (however we have not tested cellular/nuclear sub pools of this size as of 2024).
Other important points: nuclease rich cells benefit from extra RNase inhibitors at the proteinase K stage as well as on the second day of SHARE. We have increased these numbers to working with pools around 50,000-100,000 cells or nuclei, but you may need to adjust these numbers depending on your source material.
Replicates are also important: if you return 50,000 cells or nuclei for a two plate experiment, doing two cellular/nuclear sub pools of 25,000 cells is better than a single pool - it allows for concordance checking across replicates as well as providing some insurance for handling samples on the second day of SHARE.
Bring the volume of each sub-library up to 50ul using NI-2RI
For proteinase K digestion, add 50ul 2x RCB, 2ul proteinase K (20mg/mL), 2ul SUPERase RI
| A | B | C | |
| 2x RCB | Volume (mL) | 2x Concentration | |
| 1M Tris pH 8.0 | 1 | 100 mM | |
| 5M NaCl | 0.2 | 100 mM | |
| 20% SDS | 0.2 | 0.40% | |
| H2O | 8.58 | ||
| Total | 10 |
2x RCB buffer - stable for 6-12 months at room temperature.
| A | B | C | D | E | |
| ProtK mix | Volume (ul) ✕ samples (N+1) | 5 samples: N+1=6 | 10 samples N+1=11 | 20 samples N+1=21 | |
| RCB | 50 | 300 | 550 | 1050 | |
| Proteinase K (20mg/mL) | 2 | 12 | 22 | 42 | |
| SUPERase RI | 2 | 12 | 22 | 42 | |
| Cell in NI-2RI | 50 | ||||
| Total | 104 | 324 | 594 | 1134 |
Proteinase K mix volumes. Add 54ul ProtK mastermix to each sample in 50ul NI-2RI
Incubate subpools at 55ºC for one hour on the thermal cycler with lid is set to 65ºC
Can stop and store proteinase K digestions at -80ºC for up to a week. A week is definitely the maximum storage here, we've tried 2 weeks, and can't recover any cDNA (the ATAC looked fine though).
A note on terminology: we previously talked about samples - what is processed and loaded into the hybridization plate. From this point forward, we will be thinking about processing subpools - the pools of cells made in step 93 above. So when the instructions refer to "N" reactions, this now refers to the number of subpools you are processing, and not the samples you loaded on the first day of SHARE-seq.
If coming from -80ºC, thaw samples on ice first. Mix each reaction with 2.5ul 100mM PMSF in IPA and incubate at RT for 10 minutes.
To make 1mL 100mM PMSF in 100% IPA, use 17mg PMSF and weigh into a 1.5mL Eppendorf tube, mix with 1mL 100% IPA.
Prepare the following buffers:
| A | B | C | |
| 2x BW | Volume (mL) | 2x Concentration | |
| 1M Tris pH 8.0 | 0.5 | 10 mM | |
| 5M NaCl | 20 | 2 M | |
| 0.5M EDTA | 0.1 | 1 mM | |
| H2O | 29.4 | ||
| Total | 50 |
2x BW recipe - stable at room temperature
| A | B | C | |
| 1x BW-T | Volume (mL) | 1x Concentration | |
| 1M Tris pH 8.0 | 0.25 | 5 mM | |
| 5M NaCl | 10 | 1 M | |
| 0.5M EDTA | 0.05 | 0.5 mM | |
| 10% Tween 20 | 0.25 | 0.05% | |
| H2O | 39.675 | ||
| Total | 50 |
1x BW-T recipe - stable at room temperature
| A | B | C | |
| Oligo Annealing buffer (STE) | Volume (mL) | 1x Concentration | |
| 1M Tris pH 8.0 | 0.5 | 10 mM | |
| 5M NaCl | 0.5 | 50 mM | |
| 0.5 M EDTA | 0.1 | 1 mM | |
| H2O | 48.9 | ||
| Total | 50 |
STE recipe - stable at room temperature
Now prepare the above buffers with RNase inhibitors.
| A | B | |
| 2x BW/RI | Volume/ul | |
| 2x BW | 110x(N+1) | |
| SUPERase RI | 2x(N+1) |
2x BW/RI - make this fresh. Note that the RNase inhibitor concentration is adjustable - this concentration works well for 25,000-100,000 cells or nuclei.
| A | B | |
| 1x BW-T/RI | Volume/ul | |
| 1x BW-T | 500x(N+1) | |
| SUPERase RI | 10x(N+1) |
1x BW-T/RI - make this fresh. Note that the RNase inhibitor concentration is adjustable - this concentration works well for 25,000-100,000 cells or nuclei.
| A | B | |
| 1x STE/RI | Volume/ul | |
| STE | 200x(N+1) | |
| SUPERase RI | N+1 |
1x STE/RI - make this fresh. Note that the RNase inhibitor concentration is adjustable - this concentration works well for 25,000-100,000 cells or nuclei.
In a separate tube, mix 10xN ul of MyOne C1 Dynabeads with 100xN ul of 1xBW-T and place on a magnetic rack
Remove supernatant, wash twice with 100xN ul BW-T without RI
Remove supernatant, wash once with 100xN ul BW-T/RI, and resuspend beads in 110xN ul 2x BW/RI.
Add 100ul of washed beads to each sample, nutate at RT for 60 minutes
Put on a magnetic rack, transfer supernatant containing transposed ATAC chromatin fragments to a new tube for future library preparation. The supernatant is stable at RT for several hours (but we prepare the ATAC libraries right away,using a paired system where one person handles the cDNA, and the other can handle the ATAC, see step #102 below).
Wash cDNA/RNA-bound beads with 100ul 1xBW-T/RI, three times.
Place sample on magnetic rack, wash with 100ul 1xSTE/RI, without resuspending beads
9.0 RNA library preparation: template switching (time: 2h, ATAC prep during incubations)
Make the template switch mix using the recipe below:
| A | B | C | |
| Template switch mix | Volume (ul) | Final Concentration | |
| H2O | 1.25x(N+2) | ||
| 50% PEG 6000 | 15x(N+2) | 15% | |
| 5x SMART RT buffer | 10x(N+2) | 1x | |
| Ficoll PM-400 (20%) | 10x(N+2) | 4% | |
| 10 mM dNTPs, each | 5x(N+2) | 1 mM | |
| RNase inhibitor (Lucigen) | 10x(N+2) | 0.2x | |
| 100 uM TSO | 1.25x(N+2) | 2.5 mM | |
| Maxima H Minus Reverse Transcriptase (add right before RT reaction) | 2.53x(N+2) | ||
| Total | 55x(N+2) |
Template Switch Mix recipe. Note that we have doubled the amount of the lucigen RNase inhibitor to better preserve RNA.
Remove all supernatant from the beads, resuspend beads in 55ul Template switch mix, be careful to avoid beads drying out after removing supernatant
Rotate/nutate samples at RT for 30 minutes
Incubate at 42ºC for 90 minutes, shaking at 300rpm, resuspend beads at start, 30 minutes, 60 minutes, and at the end of the incubation by flicking the tubes to mix. We find RNase contamination can happen when the samples are pipetted, so we recommend mixing the settled beads by flicking for this reason.
10.0 ATAC library preparation (time: 2h50m, variable depending on qPCR results)
Using the supernatnant with ATAC fragments from step 95, purify the samples using a Qiagen MinElute PCR purification kit (Qiagen cat #: 28006) as follows:
In new tube, mix sample and 5x volume Qiagen PB buffer (1000ul), then add to MinElute spin column in two batches 2x600ul, spinning, then discarding flowthrough after each addition (as below).
Spin down the tube at 13,000rpm for 1 minute and discard the flowthrough
Add 750ul of PE Wash Buffer to the column, spin down at 13,000rpm for 1 minute, and discard flowthrough.
Spin the samples at 13,000rpm for 1 minute to capture any excess buffer.
Place the spin column into a new tube and add 11ul of EB (10mM Tris, pH 8.0) directly to the column matrix. Incubate for 1 minute.
Spin down the tube at 13,000rpm for 1 minute to elute the DNA.
Add 11ul EB to spin column, incubate for 1 minute, and spin again for total of ~20ul eluate.
Mix ~20ul sample with 29ul PCR mix (recipe below), 1ul of 25uM IDT8_i5 01-96 primer and H2O, run PCR for 5 cycles (as below)
| A | B | C | |
| ATAC PCR temp | Time | Cycles | |
| 72ºC | 5 min | 1 cycle | |
| 98ºC | 30 s | 1 cycle | |
| 98ºC | 10 s | 5 cycles ~22 min | |
| 65ºC | 30 s | ||
| 72ºC | 60 s |
ATAC initial PCR amplification program setup
| A | B | |
| ATAC PCR mix | Volume (ul) | |
| NEBnext 2x PCR mastermix | 25x(N+1) | |
| P7 primer 25uM | 1x(N+1) | |
| H2O | 3x(N+1) | |
| Total | 29ul |
ATAC PCR mastermix recipe. Don’t forget the 1ul of IDT8_i5_xx primer for each reaction!
qPCR: using 5ul sample and 10ul qPCR mix (NEBnext, Ad1.xx, P7 primer, recipe below), run the qPCR cycle program below, and calculate the number of cycles needed to each 1/3 of the plateau fluorescence (0.33 Ct). Don't forget to use a no template control (NTC) to check for PCR mastermix contamination!
| A | B | |
| ATAC qPCR mix | Volume (ul) | |
| NEBnext 2x PCR mastermix | 5x(N+2) | |
| Ad1.01 25uM | 0.2x(N+2) | |
| P7 primer 25uM | 0.2x(N+2) | |
| 10x SYBRgreen | 0.9x(N+2) | |
| H2O | 3.7x(N+2) | |
| Total | 10x(N+2) |
ATAC qPCR recipe mixture - the 10x SYBR green is made from a 10,000x stock (Thermo), and is stored in small aliquots at -20 degrees C.
| A | B | C | |
| qPCR | cycles | ||
| 98ºC | 30 s | 1 cycle | |
| 98ºC | 10 s | 20 cycles ~1 hour | |
| 65ºC | 30 s | ||
| 72ºC | 60 s |
program for qPCR cycling
example ATAC qPCR curves
We amplifed these libraries for 3 cycles - plateau around 600 RFU, 1/3 is 200, see an intersect at 3 cycles!
Save the qPCR plate to check qPCR product size on an e-gel. This can be combined with checking the cDNA after amplification. Expect a smear with an average size of 400bp. Nucleosomal banding may be present. A smaller MW band at ~270bp is likely non-nucleosomal, and does not correlate with poor quality.
An e-gel of several ATAC libraries
We recommend using an e-gel, 2%, and we run these using the ThermoFisher/Invitrogen Snap apparatus. We use the e-gel 50bp ladder.
Run additional PCR cycles for the rest of the sample (45ul), using the PCR cycle schema above, where N=0.33 Ct cycles. The number of additional cycles expected is complex and based on cell type as well as cell number. Generally libraries with a large number of cells (25000-50000+) will need only 2-4 additional cycles of amplification. Smaller numbers of cells (2000-10000) will need more like 4-10 additional cycles of amplification. These are generalities, your samples may vary, especially for non-diploid samples.
After PCR reamp, add 1ul of Exonuclease I (NEB cat # M0293L) to each library and place libraries back in thermocycler for “ExoCleanup” program (parameters below) to get rid of excess primer.
| ExoCleanup | ||
| 37ºC | 15 mins | |
| 80ºC | 15 mins | |
| 4ºC | Forever | |
| Total | ~30 min |
incubation parameters for exonuclease I digestion
Clean sample using Qiagen MinElute DNA clean up kit (225ul binding buffer, 250ul if qPCR stage was skipped), elute in 10ul EB, then repeat elution with 10ul EB, for a total of ~20ul eluate. Libraries can be stored at -20ºC or -80ºC.
11.0 cDNA amplification and cleanup (time 3h30m)
Mix each cDNA sample with 100ul H2O, magnetize and discard supernatant, wash with 200ul STE; don’t resuspend beads.
Mix 50ul PCR mix with beads, keep in existing tubes
| A | B | |
| cDNA PCR mix | Volume (ul) | |
| Kapa Hifi 2x mastermix | 25x(N+1) | |
| RNA PCR primer 25uM | 0.8x(N+1) | |
| P7 primer 25uM | 0.8x(N+1) | |
| H2O | 23.4x(N+1) | |
| Total | 50x(N+1) |
cDNA PCR amplification mix
Run PCR for 5 cycles
| A | B | C | |
| PCR | Cycles | ||
| 95ºC | 3 min | ||
| 98ºC | 20 s | 5 cycles ~32 min | |
| 65ºC | 45 s | ||
| 72ºC | 3 min |
cDNA amplification program
With samples in magnetic rack, transfer supernatant to new PCR wells.
qPCR: for each sub-library, mix 2.5ul sample with 7.5ul qPCR mix, run qPCR protocol below and calculate 0.33 Ct.
| A | B | |
| cDNA qPCR mix | Volume (ul) | |
| Kapa Hifi 2x mix | 3.75x(N+2) | |
| RNA PCR primer 25uM | 0.12x(N+2) | |
| P7 primer 25uM | 0.12x(N+2) | |
| EVAgreen 20x | 0.5x(N+2) | |
| H2O | 3.01x(N+2) | |
| Total | 7.5x(N+2) |
qPCR master mix for cDNA
| A | B | C | |
| qPCR | |||
| 95ºC | 3 min | ||
| 98ºC | 20 s | 20 cycles ~1h 38m | |
| 65ºC | 20 s | ||
| 72ºC | 3 min |
qPCR cycling conditions for cDNA
example cDNA qPCR curves
Plateau at 1000 RFU, 1/3 = 333, see an intersect at around 10 cycles.
Run additional PCR cycles for the rest of the sample (~50ul), N=0.33 Ct cycles, see step 106 above. Cycle timing for cDNA is as multifaceted as ATAC, but has the additional complexity of possible nuclease degradation. Generally, the more cells, the lower the cycle number, but this is highly variable across input material. For cultured cell lines, we often see a 50,000+ cell sample will require [6-8] additional cycles of amplification. This increases substantially for nuclei, PBMCs and hematopoietic compartment cells especially - even for samples with 50,000 cells or more, we still generally require at least 10 cycles of amplification.
Below are some generalities, but your results may vary.
for 10000 cells:
low RNA yield (like PBMCs): 14-18 cycles
medium RNA yield: 10-14 cycles
high RNA yield: 8-12 cycles
for 25000 cells:
low RNA yield (like PBMCs) 10-15 cycles
medium RNA yield (post-mortem human nuclei from colon, adrenal glands, others) 8-12 cycles
higher RNA yield (like cultured cells, mouse brain nuclei) 6-10 cycles
Run the qPCR from the cDNA on an e-gel, an example is below:
ATAC on the left and cDNA on the right (lanes 6-10). The top band in the ladder is 2.5kb
We recommend using an e-gel, 2%, and we run these using the ThermoFisher/Invitrogen Snap apparatus. We use the e-gel 50bp ladder.
Purify each sample (47.5ul) with 38ul AMpure beads (0.8x, equilibrated to RT) for cell line samples, or 28.5ul AMpure beads (0.6x) for primary samples. Elute cDNA to 20ul EB (10 mM Tris, pH 8.0). Can stop and store samples at -80ºC here. Ampure XP protocol is below, as substeps:
Bind samples and beads for 5 minutes at RT
Magnetize for 5 minutes
Wash 2x with 200ul fresh 80% ethanol with samples on magnet (~30s incubate with ethanol on beads)
Dry for ~5 minutes (can place in fume hood to speed things up). Don’t let the beads look cracked, they should look glossy, and just at the point of turning matte.
Resuspend in 20.5ul EB
Incubate 5 minutes at RT
Magnetize until clear
Pull 20ul eluate and store in a new tube at -20 degrees.
ATAC libraries are ready to sequence, cDNA requires further processing (see the next section below). This BioA and tagmentation typically occurs on a third day.
12.0 cDNA tagmentation, amplification, and indexing
Quantify cDNA concentration using BioAnalyzer using 1ul cDNA diluted 1:10 in water (or 4.5ul water and 0.5ul DNA). Ideally, dilute 50ng cDNA to 5ng/ul in ddH2O. Note: expect > 50ng cDNA, if cDNA amount is low, you can use as little as 20ng cDNA for tagmentation. Adjust the volume of DNA and H2O accordingly.
Use Diagenode Tn5 and assemble transposome by mixing a 1:100 dilution (in dilution buffer) of Tn5, dilution buffer and annealed oligo, and incubate the mix at RT for 30 minutes (see recipe below). See section 1.3 above, steps 17-21 for an overview of annealing the adapter oligos in preparation for this step. Always perform this step, and the adapter oligo annealing, on the day of tagmentation.
| Transposome | Volume (ul) | |
| 1x Tn5 | 1.25x(N+1) | |
| Dilution buffer | 1.25x(N+1) | |
| Annealed tagmentation adapter in glycerol | 2.5x(N+1) | |
| Total | 5x(N+1) |
Assembling the transposome
Mix 10ul of 5ng/ul cDNA with 10ul H2O, 25ul TD buffer (recipe below), and 5ul assembled Tn5. Incubate at 55 ºC for 5 minutes on a thermal cycler.
| 2x TD buffer: aliquot and store at -20C | Volume (ul) | 2x Final conc | |
| H2O | 7700 | ||
| 1M Tris pH 7.5 | 200 | 20mM | |
| 1M MgCl2 | 100 | 10mM | |
| Dimethylformamide (DMF) | 2000 | 20% | |
| Total | 10000 |
2X TD buffer recipe - many thanks to Rachel Savage for this homegrown recipe!
Purify tagmented library with Qiagen column (250ul binding buffer), elute twice with 11ul EB (22ul total eluate volume)
Add 29ul of post tagmentation PCR mix with 1ul tube specific (sample specific) IDT8_i5_xx primer, amplify for 7 cycles.
| A | B | |
| Post-tagmentation PCR mix | Volume (ul) | |
| Sample in EB | 20 per sample | |
| NEBnext 2x PCR mastermix | 25x(N+1) | |
| P7 primer 25uM | 1x(N+1) | |
| H2O | 3x(N+1) | |
| Total | 50x(N+1) |
Post tagmentation PCR mix. Don't forget to add 1ul of the i5 index primer!
| A | B | C | |
| Tagmentation PCR | |||
| 72ºC | 5 min | ||
| 98ºC | 30 s | ||
| 98ºC | 10 s | 7 cycles ~27 min | |
| 65ºC | 30 s | ||
| 72ºC | 60 |
tagmentation PCR cycling conditions
After PCR reamp, add 1ul of Exonuclease I to each library and place libraries back in thermocycler for “ExoCleanup” program to get rid of excess primer.
| ExoCleanup | ||
| 37ºC | 15 mins | |
| 80ºC | 15 mins | |
| 4ºC | Forever | |
| Total | ~30 min |
exonuclease cycling conditions
Purify each sample with 35ul AMpure beads (0.7x), elute library to 50ul EB, then repeat the 0.7x purification and elute in 20ul EB.
This double elution is to remove small MW fragments (less than ~250bp) which represent tagmentation events where the Illumina R2 is likely destroyed, and there is no actual cDNA retained (the SHARE scaffold takes up around 234bp). Leaving these fragments present drastically reduces the efficiency of RNA sequencing, so we highly recommend you perform these steps as written!
Bind samples and beads for 5 minutes at RT
Magnetize for 5 minutes
Wash 2x with fresh 80% ethanol with samples on magnet
Dry for ~5 minutes (can place in fume hood to speed things up). Don’t let the beads look cracked, they should look glossy.
Resuspend in 50ul EB
Incubate 5 minutes at RT
Magnetize until clear
Pull 50ul eluate and store in a new tube.
Repeat with 35ul AMpure beads, elute in ~20ul EB and store at -20 degrees C.
12.0 Library quantification and sequencing
We recommend running all of the cDNA libraries on a bioanalyzer or tapestation to assess the average molecular weight, and the presence/absence of low MW molecules (less than 250bp), which may affect sequencing quality negatively.
After confirmation of satisfactory bioanalyzer or tapestation traces, quantify all libraries to be sequenced together (in the same independently addressable lane) using a Kapa quantification kit (Roche cat number: 7960140001).
We recommend running reactions in duplicate, using 10ul reactions, loading standards in order of ascending concentration (load NTC, then std6, std5..... std1), and using two dilutions. We first dilute a library 1:1000 in water, then serially dilute 1:1000 again to get a 1:10^6 dilution. We also dilute the 1:1000 again at 1:100 to get a 1:100,000 dilution. Duplicates of both 1:10^6 and 1:100K are used to calculate the concentration of the libraries (these values will be different, that is OK).
Quantify using the Kapa Quant qPCR kit for Illumina, and pool libraries to 4-10nM concentration. Expect > 5-100nM, average library size of 600bp for cDNA, and 1-2kb for ATAC with minimal adapter dimers.
The Epstein lab typically sequences libraries on the NovaSeq X 10B, as it has the
lowest cost per base. The sequencing read configuration is as below, for separated(!!) cDNA and ATAC-seq libraries. We find separating the libraries allows for better quality sequencing data, likely due to the differences in molecular weights between cDNA and ATAC.
The recommended read structure for SHARE-seq