Oct 04, 2023
sciPlex-ATAC3
This protocol is a draft, published without a DOI.
- 1University of Washington



Protocol Citation: Gregory Booth 2023. sciPlex-ATAC3. protocols.io https://dx.doi.org/
Manuscript citation:
Booth, Gregory T., Riza M. Daza, Sanjay R. Srivatsan, José L. McFaline-Figueroa, Rula Green Gladden, Scott N. Furlan, Jay Shendure, and Cole Trapnell. 2023. “High-Capacity Sample Multiplexing for Single Cell Chromatin Accessibility Profiling.” bioRxiv. https://doi.org/10.1101/2023.03.05.531201.
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: September 13, 2023
Last Modified: October 04, 2023
Protocol Integer ID: 87763
Keywords: ATAC-seq, Chromatin, Accessibility, Gene Regulation, Nucleus, Single Cell, Genomics, High throughput screens, concurrent profiling of chromatin accessibility, chromatin accessibility, cell chromatin accessibility, chromatin fragment, epigenetic landscape of diverse tissue, atac3 assay, unmodified dna oligos as sample, epigenetic landscape, profiling cells from many independent specimen, specific nuclear label, unmodified dna oligo, sequencing library, profiling cell, dna, atac3, single nuclei, nuclei, atac, nucleus, many independent specimen, sciplex, material from each nucleus, indexing sequence
Funders Acknowledgements:
NHGRI
Grant ID: F32GM140502
Abstract
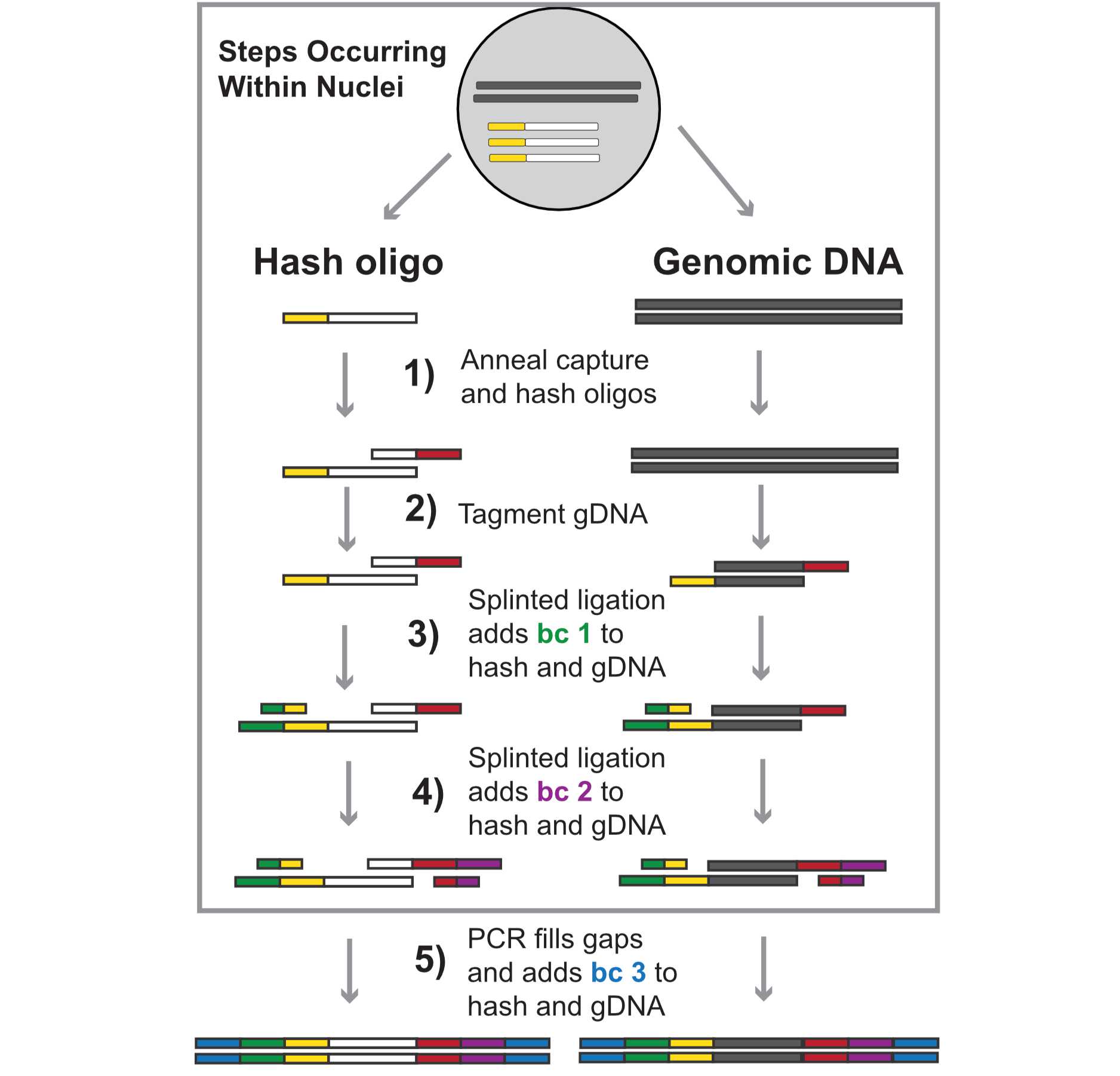
Single-cell chromatin accessibility has emerged as a powerful means of understanding the epigenetic landscape of diverse tissues and cell types, but profiling cells from many independent specimens is challenging and costly. Here we describe a novel approach, sciPlex-ATAC-seq, which uses unmodified DNA oligos as sample-specific nuclear labels, enabling the concurrent profiling of chromatin accessibility within single nuclei from virtually unlimited specimens or experimental conditions. We present a step by step protocol for the sciPlex-ATAC3 assay, where 3 layers of indexing sequences added to both hash oligos and tagmented chromatin fragments. Ultimately material from each nucleus is marked with a unique combination of indices, added through sequential split-pool reactions. This protocol describes the procedure preparing sequencing libraries using 96-well reactions (e.g. 96x96x96 barcodes), but the assay can also be scaled up (e.g. 384x384x384), depending on the desired number of unique barcodes.
Hashing and Fixing cells (cell culture)
Prepare solutions before starting:
1.5% Formaldehyde with dPBS (at least 20 mL) -- prepare fresh before fixation
0.81 mL 37% Formaldehyde
16.9 mL H20
2.5 mL 10x dPBS (final 1.25x)
ATAC-RSB buffer (Nuclei buffer) -- (50 mL) Add detergents the morning of
10mM TrisHCl
10mM NaCl
3mM MgCl2
Separately prepare 5 mL with added 0.1% Tween
OMNI lysis buffer -- Add detergents the morning of (OMNI ATAC paper = PMID: 28846090)
10mM TrisHCl
10mM NaCl
3mM MgCl2
0.1% NP40 --Add Fresh
0.1% Tween --Add Fresh
0.01% Digitonin --Add Fresh
1x Protease inhibitor (Thermo Pierce‱ Protease Inhibitor Tablets, EDTA-free) --Add Fresh
Freezing Buffer Base (prepare 50 mL for storage at 4C up to 6 months)
50 mM Tris pH 8.0,
25% glycerol,
5 mM Mg(OAc)2,
0.1 mM EDTA,
H20
Filter Sterilize (22um)
Freezing Buffer (working) (1mL)
975 uL Freezing Buffer Base
5 uL 1M DTT
20 uL protease inhibitor cocktail (Thermo Pierce‱ Protease Inhibitor Tablets, EDTA-free)
- Spin down culture plate (300xG at for 5 minutes room temperature) (e.g. Cells grown in 96-well format), remove culture media and resuspend in 200uL 1xDPBS.
- Spin down and remove 1xDPBS (300XG at for 5 mins room temperature).
- If using adherent cells: add 50uL of Tryp-LE per well and place plate into a 37C incubator and allow to incubate for 5-10 minutes based on the cell line.
- After cells are detached, add 150uL of culture medium to quench the Tryp-LE.
- Using a new set of tips for every well and a multichannel pipette, transfer the 200uL volume cell suspension into a V-bottom 96 well plate. Make sure that the orientation is preserved between the 96 well culture plate and the 96 well V-bottom plate.
- Spin down again at 300xG for 5 minutes, remove media by aspiration.
- Add 100uL of cold 1xDPBS with a multichannel to every well to wash off residual media.
- Spin cells down again at 300xG for 5 minutes at 4C in the V-bottom plate to pellet cells and remove supernatant.
- Resuspend each well in 50uL ice cold OMNI lysis buffer and mix several times with widebore tips.
- Add 1uL of distinct hash oligos (10uM) to each well and incubate on ice for 5 mins.
- Note: We've found that hashing efficiency can decrease if too little oligos are used. It is best to consider the starting number of cells within each well and target 0.5pmol hash molecules per 1000 cells.
- Hashes have the sequence 5’GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGXXXXXXXXXXBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA-3’, where ‘X’s represent the 10nt, well-specific hash ID.
- Add 102 uL 1.5% Formaldehyde to each well for FC = 1% and mix with widebore tip.
- Allow fixation to occur on ice for 15 minutes.
- Hashed nuclei can now be pooled. Combine nuclei (in fixation solution) from all wells into a 15mL conical.
- Spin down at 600xG for 10 minutes at 4C.
- Resuspend in 1mL of ice cold Nuclei Buffer with 0.1% tween20
- Allow to rest on ice for 3 minutes.
- Spin down at 600g for 5 minutes at 4C.
- Resuspend in 0.5 mL Freezing buffer.
- Count cells and bring to final conctration of approx. 5x10e6 cells/mL in freezing buffer (working sln).
- Flash freeze and stored at -80C. Otherwise proceed directly to next section.
- Note: Freeze thaw may contribute to nuclear lysis, so it is ideal to proceed without freezing when possible; however, the next freeze step is following both ligations and PCR.
Hash capture oligo annealing ~ 15 Minutes
- Distribute 50,000 nuclei/well (10 uL of 5x10e6/ml) to desired number of wells.
- Note: Typically we will load 24 wells (1.2x10e6 cells) for 96x96x96 well barcoding experiments. For 384x384x384 experiments we will load 96 wells (4.2x10e6 cells)
- Add 2 uL of 25uM PolyDT (LNA) capture oligo + UMI.
- Hash Capture Oligo Sequence:
- 5’TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGNNNNNNNNXXXXXXXXXXTTT+TTT+TTT+TTT+TTT+TTT+TTT+TTT+TTT+TTTVN-3’ Where ‘X’s represent a well specific barcode while ‘N’s reflect the unique molecular index (UMI). ‘+’ indicates the next nucleotide is a locked nucleic acid (LNA). The locked nucleic acids placed every 3rd position increases the affinity for PolyA hash molecules and is critical to ensure the fidelity of hash capture.
- Incubate plate at 55C for 5 minutes using a thermal cycler (total volume 12uL).
- Return to ice and let cool for 5 minutes.
Tagmentation of accessible chromatin ~
Prepare Tn5 reaction and transfer 35.5 uL of the Tn5 reaction mix (lacking Tn5) to each well.
| Reagent | volume (1 well) | volume (110 wells) | |
| TD buffer 2X | 25uL | 2750uL | |
| 1x DPBS | 8.25uL | 907.5uL | |
| 1%Digitonin | 0.5uL | 55uL | |
| 10%Tween20 | 0.5uL | 55uL | |
| Water | 1.25uL | 137.5uL |
- Using a multi-channel, add 2.5ul of Tn5 (Illumina/Nextera) to each well. Total well volume is now 50ul.
- Seal with an adhesive tape then spin at 500xG for 30 sec.
- Incubate at 55C for 30 minutes using a thermal cycle.
- NOTE: During tagmentation incubation, prepare stop reaction (EDTA/spermidine) mixture.
| Reagent | Vol | |
| 1mM spermidine | 0.78uL | |
| 40mM EDTA | 5mL |
- Add 50ul of ice cold EDTA/Spermidine mixture to each well using a wide bore tip.
- NOTE: Default to normal tips to increase capture efficiency; however, at certain steps where nuclei are fragile, the protocol will suggest using wide bore tips.
- Incubate at 37°C for 15 minutes using a thermal cycle.
- Using a wide bore tip, separately pool each well from each well into a 15ml conical tube.
- Pellet tagmented nuclei at 600xG for 10minutes at 4°C and aspirate supernatant.
- Wash with 500 ul RSB-ATAC + 0.1% Tween (carefully resuspend the nuclei).
- Spin at 600xG for 5 min at 4°C.
- Aspirate supernatant and resuspend pellet in 110 ul RSB-ATAC+0.1% Tween.
PNK treatment of ATAC fragment ends ~ 1 hour
- Make a PNK master mix (prepare 110 volumes for 96well plate).
| Reagent | 1x Reaction vol | 110x Reaction vol | |
| 10x PNK buffer | 0.5uL | 55uL | |
| rATP 10 mM | 0.5uL | 55uL | |
| water | 1uL | 110uL | |
| T4 PNK | 2uL | 220uL |
- Add 440uL of master mix to 110uL of nuclei for a 96 well plate.
- Aliquot 5.5uL of master mix and tagmented cells to a new 96-well plate.
- Seal with an adhesive tape then spin at 500xg for 30 seconds at 4C.
- Incubate at 37°C for 30 min using a thermal cycler.
Barcode Ligation (Round 1 barcode) (N7 side) ~ 1.5 hours
- Prepare the following master mix (without N7 oligos) and add 12.86 ul of ligation master mix directly to PNK reaction within 96 wells.
| Reagent | Vol. per well | |
| 2X T7 ligase buffer | 10 | |
| 1000 uM N7_splint | 0.18 | |
| H20 | 1.38 | |
| T7 DNA ligase | 2.5 | |
| 50 uM N7_oligo | 1.2 |
- Prepare the following master mix (without N7 oligos) and add 12.86 ul of ligation master mix directly to PNK reaction within 96 wells.
- N7_splint oligo has the sequence: 5’-CACGAGACGACAAGT-3’
- Add 1.2 ul of N7_oligo (50 uM) to each well.
- Note: each well should receive a unique N7_oligo index.
- N7_oligo has the sequence: 5’-CAGCACGGCGAGACTNNNNNNNNNNGACTTGTC-3’ , where ‘N’s represent a well specific index).
- Seal with an adhesive tape then spin at 500xg for 30 seconds at 4C.
- Incubate at 25C for 1 hour in thermal cycler.
- NOTE: During ligation incubation, prepare stop reaction mixture.
| Reagent | Vol | |
| 40mM EDTA | 5mL | |
| 1mM spermidine | 0.78uL |
- Add 20 ul of EDTA/Spermidine mixture using a wide bore tip.
- Incubate at 37°C for 15 min using a thermal cycler.
- Using a wide bore tip, pool each well into a 15ml conical tube.
- Add 3X volumes RSB-ATAC buffer + 0.1% tween20 and pellet nuclei by spinning at 600xG for 10 min. Remove supernatant.
- Resuspend nuclei in RSB-ATAC buffer + 0.1% tween20 (Volume for 5uL per well in subsequent ligation, e.g. 550uL for 96 wells), gently pipet up and down.
Barcode Ligation (Round 2 barcode) (N5 side) ~ 1.5 hours
- Distribute 5.5uL of resuspended nuclei to each well of a new 96 well plate.
- Prepare ligation mix (lacking n5 oligo) and add entire mix directly to pooled nuclei (accounting for total pool volume).
| A | Volume (1 well) | |
| 2X T7 ligase buffer | 10uL | |
| 1000uM N5_splint | 0.2uL | |
| H20 | 1.1uL | |
| T7 DNA ligase | 2.5uL | |
| 50uM N5_oligo | 1.2uL |
- The N5_splint oligo has the sequence: 5’-GCCGACGACTGATTA-3’
- Distribute 18.8 uL of mix (containing nuclei) to each well of new 96 well plate.
- Add 1.2 ul of N5_oligo_50 uM to each well.
- Note: each well should receive a unique N5_oligo index.
- N5_oligo has the sequence: 5’-CACCGCACGAGAGGTNNNNNNNNNNGTAATCAG-3' , where ‘N’s represent a well specific index).
- Seal with an adhesive tape then spin at 500xg for 30 sec.
- Incubate at room temperature (25C) for 1 hour in thermal cycler.
- Gently add 20uL of Stop Reaction Mix (EDTA/Spermidine) using a wide bore tip.
| A | B | |
| 40mM EDTA | 5mL | |
| 1mM spermidine | 0.78uL |
- Incubate at 37°C for 15 min on thermal cycler.
- Using a wide bore tip, pool each well into a 15ml conical tube.
- Add 3X volumes EB buffer (Qiagen) and pellet nuclei by spinning at 600xG for 10 min. Remove supernatant.
- Resuspend nuclei in 0.5 ml EB buffer (Qiagen), gently pipet up and down.
Final Dilution and Reverse X-links ~ Overnight
14h
- Determine nuclei concentration using trypan blue staining, then dilute nuclei to desired concentration and distribute 10 uL into a new 96 well Lo-bind plate for PCR.
- Example: when using 96 ligation indices each round (96x96x96 format) you can target 1000 cells/well (dilute to 100,000 nuclei /mL) and the expected doublet rate will be ~10%.
- Make a REV-X-Link master mix, add 2 ul to each well.
| Reagent | Volume (1 well) | |
| Qiagen EB buffer | 1uL | |
| Proteinase K (20mg/uL) | 0.5uL | |
| 1% SDS | 0.5uL |
- Seal with an adhesive tape then spin at 500xg for 30 sec.
- Uncrosslink at 65°C for 13.5 hours or overnight using a thermal cycler.
- If not proceeding directly to PCR, diluted nuclei in plates can be stored at -20°C.
PCR amplification (Round 3 barcode) ~ 2 hours
- Quick centrifuge of plates plates after uncrosslinking.
- Prepare a PCR Mix (2X NEB NEXT, Water, BSA) lacking primers and add 33uL to each well with nuclei.
| A | B | |
| Sorted nuclei (25 nuclei/well) | 12.0 | |
| P7_primer | 2.5 | |
| P5_primer | 2.5 | |
| NEBNext High Fidelity 2x PCR Master Mix | 25 | |
| Water | 7 | |
| BSA (100x) | 1 |
- P7 primer: 5’-CAAGCAGAAGACGGCATACGAGATNNNNNNNNNNCAGCACGGCGAGACT-3’
- P5 primer: 5’-AATGATACGGCGACCACCGAGATCTACACNNNNNNNNNNCACCGCACGAGAGGT-3’
- "N"s represent well specific barcodes. The combination of these two barcodes comprises the 3rd level index.
- Seal with an adhesive tape then spin at 500xg for 30 sec.
- Run PCR plate with between 20-25 cycles:
72ºC 5 min
98ºC 30 sec
20 Cycles (or choose optimal):
98ºC 10 sec
63ºC 30 sec
72ºC 1 min
Hold at 10ºC
- Note: Optimal cycle number is determined by using qPCR (on a subset of wells) to determine the number of PCR cycles at which curves begin to saturate. Libraries which require more than 25 cycle will likely have poor overall quality.
Purify final library and get fragment size profile ~ 1.5 hours
Use the Zymo Clean&Concentrator-5 kit to purify pooled libraries.
- Combine 25ul of each PCR reaction (2.4ml from 96 well plate) to a trough.
- Note: This is half of the volume from the PCR plates. The other half can be saved at 4C or -20C for later sequencing/troubleshooting as needed.
- Add 2 volumes binding buffer (4.8ml).
- Split across 4 Clean and Concetrate columns (600uL spun 3 times in each column).
- Use an extra spin to dry columns for 1min after washes.
- Elute in 25ul Qiagen elution buffer in each tube (let buffer stand on column 1min, then spin 1min).
- Pool the clean and concentrate eluates for a final volume of 100uL.
- Determine library concentration by Qubit and fragment size profile by Screen tape(D5000).
- OPTIONAL: Can perform additional SPRI cleanup if primer dimers (~150bp) make up a significant potion of library fragments. However, extra size selection might contribute to hash loss, so be careful.
Sequencing the Library ~ 1.5 hours
Libraries can be assessed for quality with a NextSeq run before deeper sequencing.
- Dilute 2M NaOH to 0.2M NaOH (10uL 1M to 90uL nuclease-free water).
- Add 10uL 0.1M NaOH and add 10uL 2nM pooled libraries.
- Incubate at RT for 5 minutes.
- Add 980uL HT1 to dilute denatured libraries to 20pM.
- Dilute denatured library to 1.8pM loading concentration (135uL 20pM + 1365uL HT1).
- Dilute custom primers for seqeuncing to 0.6uM.
- Custom Primer Positions to Add on a NextSeq 550 kit in positions
| Position on Kit | ID | Volume of 100uM Primer | Volume of HT1 | Volume to Load In Kit | |
| 7 | Read 1 | 9uL | 1491uL | 1500uL | |
| 8 | Read 2 | 9uL | 1491uL | 1500uL | |
| 9 | Index 1 & Index 2 | 9uL + 9uL | 1491uL + 1491uL | 3000uL |
- NextSeq Sequencing Recipe uses custom light & dark cycles.
R1 - 50 bases for gDNA.
R2 - 50 bases for gDNA.
Index 1 - 20 bases (10 bases for N7 oligo, 15 dark cycle, 10 bases PCR barcode).
Index 2 - 20 bases (10 bases for N5 oligo, 15 dark cycle, 10 bases PCR barcode).
- The sequencing primers, different from the default Illumina primers, are below.
| A | B | |
| 3Lv2_R1_seq | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG | |
| 3Lv2_R2_seq | GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG | |
| 3Lv2_IDX1 | CTCCGAGCCCACGAGACGACAAGTC | |
| 3Lv2_IDX2 | ACACATCTGACGCTGCCGACGACTGATTAC |
Protocol references
Booth, Gregory T., Riza M. Daza, Sanjay R. Srivatsan, José L. McFaline-Figueroa, Rula Green Gladden, Scott N. Furlan, Jay Shendure, and Cole Trapnell. 2023. “High-Capacity Sample Multiplexing for Single Cell Chromatin Accessibility Profiling.” bioRxiv. https://doi.org/10.1101/2023.03.05.531201.
Domcke, Silvia, Andrew J. Hill, Riza M. Daza, Junyue Cao, Diana R. O’Day, Hannah A. Pliner, Kimberly A. Aldinger, et al. 2020. “A Human Cell Atlas of Fetal Chromatin Accessibility.” Science 370 (6518). https://doi.org/10.1126/science.aba7612.
Srivatsan, Sanjay R., José L. McFaline-Figueroa, Vijay Ramani, Lauren Saunders, Junyue Cao, Jonathan Packer, Hannah A. Pliner, et al. 2020. “Massively Multiplex Chemical Transcriptomics at Single-Cell Resolution.” Science 367 (6473): 45–51