Apr 23, 2024
SARS-CoV-2 incursion scenario in the city Fantastica v2
- 1Australian National University



Protocol Citation: Benjamin Schwessinger 2024. SARS-CoV-2 incursion scenario in the city Fantastica v2. protocols.io https://dx.doi.org/10.17504/protocols.io.kqdg326xpv25/v1
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: April 23, 2024
Last Modified: April 23, 2024
Protocol Integer ID: 98644
Keywords: genomic surveillance of sar, following about sar, spread into of sar, spread of sar, genomic surveillance, likely infection scenario for the family infection cluster, family infection cluster, classic epidemiological data with genomic data, sar, available sar, australia on sar, official sar, known outbreak location, exclusive genomic surveillance, genome per infection cycle, pathogen, biosecurity sector, outbreak reference id area, cov2, outbreak reference id area of residence age date, weakness of exclusive genomic surveillance, genomic, combining classic epidemiological data, human biosecurity, used sar, outbreak reference id, genomic data, potential exposure vaccination status oversea, tutorials around human biosecurity, likely infection scenario, quarantine, fantasticasarscov2sequence, matching genomic dataset, genome, viral mutation rate, studying incursion scenario, cov, hospitalcluster1, likely source of infection, vaccination coverage, incursion scenario, infection cycle, city fantastica v2, quarantin
Abstract
This protocol is part of the ANU Biosecurity mini-research project #2 "An SARS-CoV-2 incursion scenario: Genomics, phylogenetics, and incursions." This mini-research project aims to highlight the power of combining classic epidemiological data with genomic data of the pathogen when studying incursion scenarios. This mini-research project is modeled on the yearly Quality Assurance Program (QAP) of The Royal College of Pathologists of Australia on SARS-CoV2, which we used to complete in collaboration with ACT Health.
This research project is has a single component from 2024 onward based on student feedback in the previous years. It focuses on the 'dry-lab' by investigating a hypothetical incursion scenario in the so-called city Fantastica. You will combine genomic surveillance of SARS-CoV-2 with case interview data to trace the spread into of SARS-CoV-2 in the community and into high risk settings. We will provide you with real publicly available SARS-CoV-2 genome and fantasized case interviews. You will put these two together to trace the spread and suggest potential improvements in containment strategies with a focus on high risk settings.
This is a creative version of similar scenarios investigated during the official SARS-CoV-2 QAPs. The main objective of mini-research project #2 is to solidify concepts you learned in the lectures and tutorials around human biosecurity. We will combine fictional case interview information with a matching genomic dataset of SARS-CoV-2 genomes to investigate the incursion. Hopefully this will show you the power combining these two data types brings when compared to having only one or the other. In the larger perspective of the course, this hopefully illustrates to you that one needs to consider a multitude of perspectives and data types when operating in the biosecurity sector.
I had a lot of fun coming up with this incursion scenario and I hope you will enjoy working on it with your detective hat on. Of course this complete scenario is absolutely fictional. All the used SARS-CoV-2 sequences are publicly available on GISAID as described in this publication (Hall et. al. 2023).
The incursion scenario:
Imagine a city called Fantastica in the middle of the SARS-CoV-2 pandemic mid-2021 in a country where vaccination coverage and COVID-19 case numbers are very low. Fantastica is located on a continental scale island nation and the international borders to this nation are highly regulated to prevent new COVID-19 cases from entering. The main public health measures employed to contain the spread of SARS-CoV-2 are social distancing, mask wearing, mass testing, contact tracing, isolating and quarantining of confirmed cases and lock-downs.
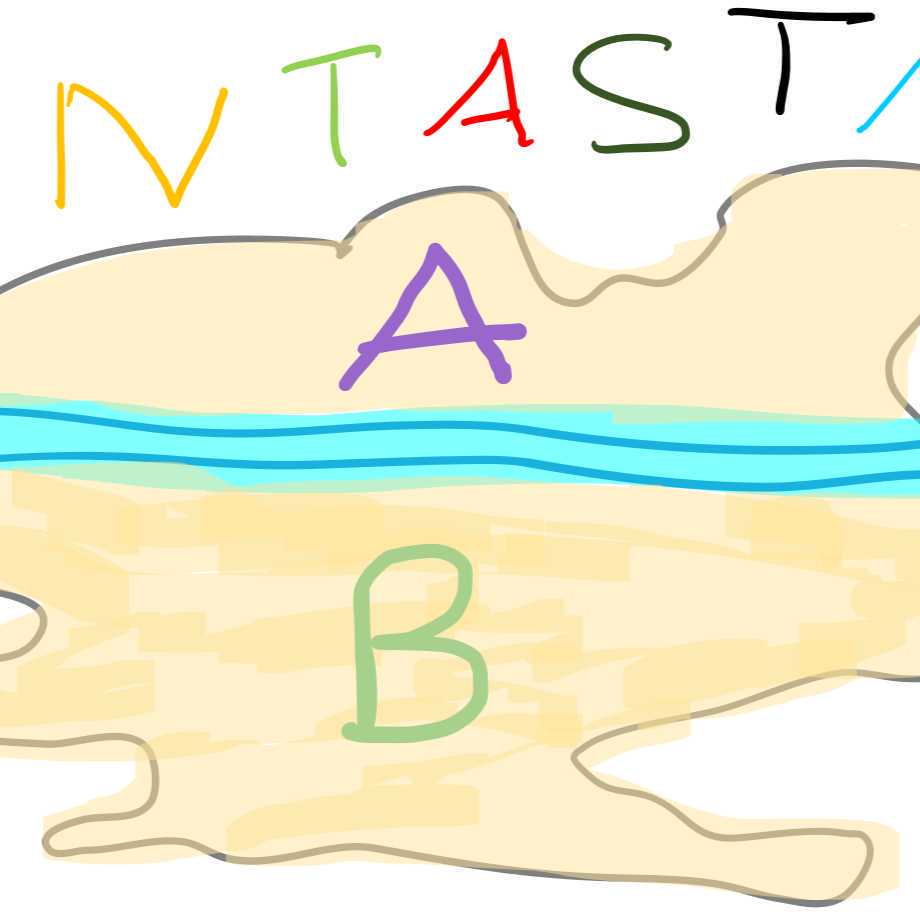
Fantastica has two main areas of residence with A being the affluent North and B being the less well off South (Figure 1). These two areas are separated by a river. The main hospital is located right at the river.
City map Fantastica
In mid-2021 the city experiences its first COVID-19 case for a long time (Outbreak reference ID: Fantastica034), which was successfully contained in hotel quarantine for overseas travelers. The following months Fantastica experiences a larger COVID-19 outbreak that it aims to contain with lockdowns including restricting movements from 12 September 2021 till 20 November 2021. The public health unit achieves to sequence all SARS-CoV-2 genomes of all identified COVID-19 cases in this time frame.
In our simplified scenario we assume the following about SARS-CoV-2:
- Infectious period: 48 hrs before and after onset of symptoms.
- Asymptomatic cases can also cause forward transmission.
- Viral mutation rate: on average 0.5 mutations in each genome per infection cycle.
You are now provided with the following material to start your investigation and address the specific questions below. All the information is idealized and fictionalized.
1. An excel file (ContactTracingCaseInterviews) containing case interview information (not exhaustive and simplified) including the following columns:
- Outbreak Reference ID
- Area of Residence
- Age
- Date of symptom onset
- Date of specimen collection
- Symptoms
- Household contact
- Contact with known COVID-19 case
- Case associated with known outbreak
- Locations of potential exposure
- Vaccination Status
- Overseas travel
2. A fasta file (FantasticaSARSCoV2Sequences) of SARS-CoV-2 genomes of all identified COVID-19 cases in Fantastica in the indicated study period (plus Fantastica034)
3. A PNG file (CleanedUpAlignmentAllSequencesTree) of the simple Neighbor-joining tree. You will generate the same tree in class.
What you need for the prac:
- A detective hat.
- Your computer.
- Pen and paper including different colored pens.
Specific questions to be addressed in the prac and your final report:
- Describe the overall LargeClusterA1. What drove the transmission in this cluster? Was it contained successfully with public health measures such as testing, tracing, lockdowns and quarantine? Has the index case been clearly identified? Is the index case the likely first case in this cluster? Do you think most cases in this cluster have been identified? Explain your reasoning.
- Describe the overall LargeClusterB1. What drove the transmission in this cluster? Was it contained successfully with public health measures such as testing, tracing, lockdowns and quarantine? Has the index case been clearly identified? Is the index case the likely first case in this cluster? Do you think most cases in this cluster have been identified? Include later appearing mini-clusters in your analysis:
MC1: Fantastica063, Fantastica062, Fantastica058, Fantastica064 and Fantastica059.
MC2: Fantastica067, Fantastica068, Fantastica069, Fantastica072, Fantastica074, Fantastica070, Fantastica071, Fantastica073, Fantastica075
In your analysis speculate how, these genetically linked subclusters could potentially physically linked (or not) to the main cluster?
- What is a likely infection scenario for the family infection cluster containing Fantastica014, 016, 017?
- How can you explain that case Fantastica019 is so distinct from all other cases?
- Describe the case Fantastica033. What cluster does this case belong to? When could this case have caught COVID-19? Who could be the potential source cases? Where could this case got infected? Explain your reasoning.
- Describe the "HospitalCluster1 (non-COVID Ward)"? Was it a single incursion? What was the likely transmission chain? How could such an incursion scenario be better managed? Explain your reasoning.
- Describe the "ElderlyHomeClusterB"? Was it a single incursion? What was the likely transmission chain? How could such an incursion scenario be better managed? Explain your reasoning.
- How would you have interpreted case Fantastica076 without contact tracing data? What does this case reveal for the strength and weakness of exclusive genomic surveillance without epi data?
- There is one case that lied on the contact tracing form. Identify this case, its most likely source of infection, and who they passed it on to.
For all these questions we are looking for the most parsimonious answers. The simplest and most plausible answers.
Guidelines
You must have read, understood, and follow the health and safety instructions provided in the "Overview Mini-Research Project #2 BIOL3106/6106" provided on Wattle (ANU learning portal).
You must have signed and returned one copy of the "Student Safety Declaration Form For Practical Class Work" before starting any laboratory work.
Safety warnings
This protocol does not require any hazardous substances or infectious agents. However, maintain a proper posture while working on your computer.
Before start
You must study the protocol carefully before you start. If anything is unclear post questions directly here on protocols.io.
Section I: Setup Genenious and import files into Geneious
Open up Geneious.
First we have to install the MAFFT alignment plugin. Go to "Tools > Plugin"
Select to install MAFFT. This will take only a 1-2 mins to install.
Now create a folder with right click in the left folder sidebar. Call this folder something meaning full e.g. BIOL3106. Make another folder for the initial analysis of all genomes e.g. InClass. You can use a folder structure to investigate your different outbreaks later on.
Now you import all the data needed in Geneious. Track and drop the "FantasticaSARSCoV2Sequences" and the reference sequence "MN908947.3" into this in class folder.
You are ready for all your analysis now.
Section II: Generation of a multiple sequence alignment in Geneious
Great well done to set it all up. you are ready to generate your first whole genome alignment.
Now you will select all sequences and generate a multiple sequence alignment with MAFFT.
Now you have generated your first alignment. This aligns each base of all the genomes you selected to each other. I suggest you rename this alignment from "Nucleotide alignment" to something more meaningful.
With this specific display "Highlighting" setting "Disagreements" each of the black bars is a variation (mutation) compared to the consensus sequence.
Regions in the consensus sequence highlighted as red are not well covered in the aligned genomes. You can visualise this more when changing the "Highlighting" settings to something else. Play around and ask questions in class.
Section III: Building a very simple Neighbor-joining tree
Now you will build a very simple Neighbor-joining tree. If you want to learn more about tree building and phylogenetics I suggest you pick other 3rd year ANU biology courses.
Right click on your sequence alignment and select "Tree..."
Now you have your first overall tree.
The last thing for now that you need to do is the root your tree on the reference sequence "MN908947.3".
Select the "MN908947.3" by clicking on it and hit the "Root" bottom.
You have your first tree of all the sequences rooted with the original SARS-CoV-2 sequence. You can now overlay the case interview information to answer the questions for this part of the mini-research project #2. We will step through those in class as well. We will also explain how to interpret trees in more detail in class.
Importantly, you can generate these simple trees for subclusters as well (e.g. Hospital) if needed to address the questions better. This will be done by only selecting the sequences of interest for the alignment and tree building. Make sure to always include the reference and to root your tree on it.
Section IV: Overlay case interview information on top of the genetic data
So you have a skeleton (aka tree) of the genetic relationship of all samples and hence COVID-19 cases. We will provide you with a large print out copy as well.
Now you have to overlay the case interview data to answer the specific questions in the description section of the protocol above. For this you can use the printed trees to draw on (with different coloured pens) or annotate it on your computer. Make sure to make good use of the sort and filter functions in Excel when going over the case interview data to ease your analysis.
^^Screenshot of case interview data.
We will walk through some of these questions from the description section in class and will be guided by your questions.