Jan 28, 2025
RNA Extraction and RNA-Sequencing Method for Transcriptomic Analysis of Mycobacterium tuberculosis
- Morgan Hiebert1,2,
- Meenu Sharma1,2,
- Alwyn Go3,
- Christine Bonner3,
- Vanessa Laminman3,
- Morag Graham3,2,
- Hafid Soualhine4
- 1National Reference Centre for Mycobacteriology, National Microbiology Laboratory Branch, Public Health Agency of Canada;
- 2Department of Medical Microbiology and Infectious Diseases, University of Manitoba;
- 3Genomics Core Facility, National Microbiology Laboratory Branch, Public Health Agency of Canada;
- 4National Microbiology laboratory Branch, Public health Agency of Canada
- BioTechniques



Protocol Citation: Morgan Hiebert, Meenu Sharma, Alwyn Go, Christine Bonner, Vanessa Laminman, Morag Graham, Hafid Soualhine 2025. RNA Extraction and RNA-Sequencing Method for Transcriptomic Analysis of Mycobacterium tuberculosis. protocols.io https://dx.doi.org/10.17504/protocols.io.3byl49dqrgo5/v1
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: August 08, 2024
Last Modified: January 28, 2025
Protocol Integer ID: 104989
Keywords: Mycobacterium tuberculosis, RNA extraction, RNA-sequencing, Transcriptomics, Next-generation sequencing, seq of mycobacterium tuberculosis, mycobacterial transcriptomic, important gap in mycobacterial transcriptomic, mycobacterium tuberculosis, rna extraction, including rna extraction, methodology for rna, computational protocol for rna, rna, application of rna, transcriptomic analysis, method for transcriptomic analysis, mycobacterium, cdna library preparation, throughput sequencing, cdna library, rrna depletion, sequencing method, sequencing, differential expression analysis, seq
Funders Acknowledgements:
Genomics Research and Development Initiative
Grant ID: GRDI-07
Research Manitoba Masters Studentship
Grant ID: MH
Abstract
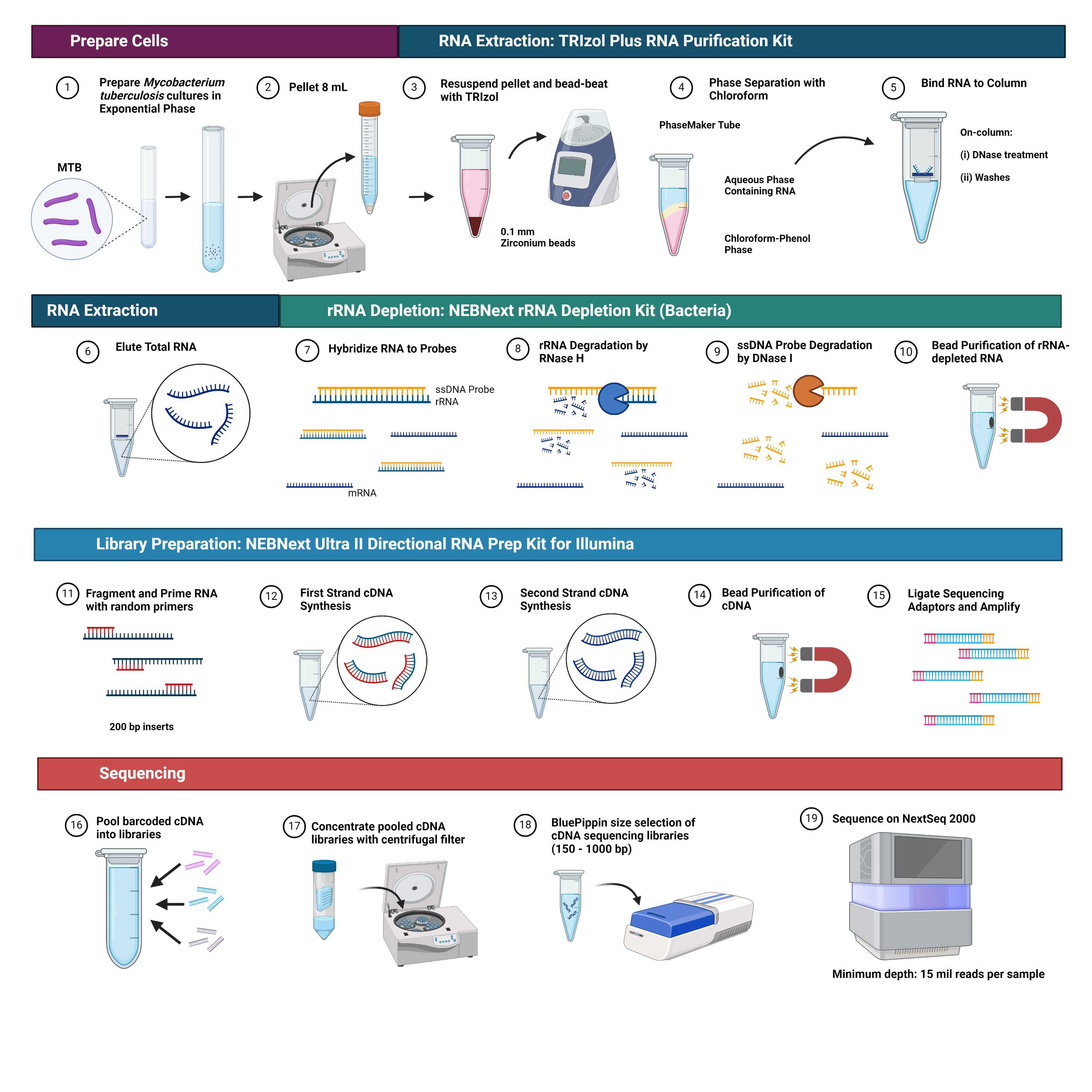
RNA-sequencing (RNA-seq) technologies have advanced exponentially in recent years, however application of RNA-seq to M. tuberculosis remains limited. We present a complete wet-lab and computational protocol for RNA-seq of Mycobacterium tuberculosis including RNA extraction, rRNA depletion, cDNA library preparation, high-throughput sequencing, and differential expression analysis. This research contributes to the literature by providing methodology for RNA-seq of M. tuberculosis, thereby bridging an important gap in mycobacterial transcriptomics.
Attachments

Image Attribution
Graphical abstract created with BioRender.com
Guidelines
This protocol requires pure cultures of Mycobacterium tuberculosis, a Risk Group-3 (RG-3) organism, and should be carried out according to regional and institutional biosafety guidelines, such as the Biosafety Directive for Mycobacterium tuberculosis Complex [1]. The protocol contains variations on the manufacturer's protocols for the TRIzol Plus RNA Purification Kit and Phasemaker Tubes Complete System [2], NEBNext rRNA Depletion Kit (Bacteria) with RNA Sample Purification Beads and the NEBNext‱ Ultra‱ II Directional RNA Library Prep kit with Sample Purification Beads [3].
Materials
Supplies
1. 0.9 McFarland Standard (E1039; Thermo Fisher Scientific, MA, USA)
2. 1.5% Agarose dye-free Gel Casette with internal standards, 250 bp – 1.5 kb (BDF1510; Sage Science, MA, USA)
3. Agilent RNA 6000 Nano Kit (5067-1511; Agilent Technologies, CA, USA)
4. Amicon‱ Ultra-0.5 Centrifugal Filter Units (UFC505096; MilliporeSigma, MA, USA)
5. BD BACTEC‱ MGIT‱ 960 PZA Kit (245128; Becton Dickinson, NJ, USA), or BD BACTEC‱ MGIT‱ 960 SIRE Kit (245128; Becton Dickinson), or BD BACTEC‱ MGIT‱ 960 Supplement Kit (245124, Becton Dickinson)
6. BD BACTEC‱ MGIT‱ 960 PZA Medium (245115; Becton Dickinson) or BD BACTEC‱ MGIT‱ Mycobacterial Growth Indicator Tubes (245122, Becton Dickinson)
7. Chloroform (34854; MilliporeSigma)
8. Ethanol (P016EA95; Commercial Alcohols Incorporated, ON, CA)
9. Falcon‱ 15 mL Conical Centrifuge tubes (05-527-90; Thermo Fisher Scientific)
10. Flip cap Microcentrifuge Tubes: 2.0 mL (05-408-141; Thermo Fisher Scientific)
11. Genomic DNA ScreenTape and Reagents (5067-5366; 5067-5365; Agilent Technologies)
12. Microcentrifuge Tubes with Screw Caps: 2 mL (02-681-375; Thermo Fisher Scientific)
13. NEBNext‱ Multiplex Oligos for Illumina‱ (96 Unique Dual Index Primer Pairs) (E6440S; New England Biolabs, MA, USA)
14. NEBNext‱ rRNA Depletion Kit (Bacteria) with RNA Sample Purification Beads (E7860X; New England Biolabs)
15. NEBNext‱ Ultra‱ II Directional RNA Library Prep kit with Sample Purification Beads (E7765L; New England Biolabs)
16. NextSeq‱ 1000/2000 P2 300M Reagents (600 cycles) (20075295; Illumina, CA, USA)
17. PureLink‱ DNase Set (12185010; Thermo Fisher Scientific)
18. Qubit‱ dsDNA High Sensitivity Assay kit (Q32854; Thermo Fisher Scientific)
19. Qubit‱ RNA High Sensitivity Assay kit (Q32852; Thermo Fisher Scientific)
20. RNaseZap‱ RNase Decontamination Solution (AM9780; Invitrogen, MA, USA)
21. Triple-Pure‱ 0.1 mm zirconium beads (D1132-01TP; Benchmark Scientific, NJ, USA)
22. TRIzol‱ Plus RNA Purification Kit and Phasemaker‱ Tubes Complete System (A33254; Thermo Fisher Scientific)
23. UltraPure‱ DNase/RNase-free Distilled Water (10977035; Invitrogen)
Equipment
1. 4200 TapeStation System (G2991BA; Agilent Technologies)
2. Agilent 2100 Bioanalyzer System (G2938A; Agilent Technologies)
3. BD BACTEC‱ MGIT‱ 960 automated mycobacterial detection system (445885; Becton Dickinson)
4. BluePippin Size Selection System (BLU0001; Sage Science)
5. Fastprep-24‱ Classic bead beating grinder and lysis system (116004500; MP Biomedicals, CA, USA)
6. Labnet Orbit‱ 300 Multipurpose Digital Shaker (S2030-300-B; Mandel Scientific Company Inc, ON, CA).
7. MiniSpin‱ Mini Centrifuge (022620100; Eppendorf, Hamburg, Germany)
8. NanoDrop‱ 2000 spectrophotometer (ND-2000; Thermo Fisher Scientific)
9. NEBNext‱ Magnetic Separation Rack (S1515S; New England Biolabs)
10. NextSeq 2000 System (Illumina)
11. Qubit‱ 4.0 Fluorometer (Q33238, Invitrogen)
12. Sorvall‱ ST 16R Centrifuge (75004240; Thermo Fisher Scientific)
13. Thermocycler
14. VWR‱ Analog Vortex Mixer (10153-838; Avantor, PA, USA)
Troubleshooting
Safety warnings
Biohazard Warnings: Mycobacterium tuberculosis is a risk group 3 (RG-3) organism and a Biosafety Level 3 (BSL-3) laboratory is required for higher risk research and diagnostics activities, as specified in the Biosafety Directive for Mycobacterium tuberculosis Complex [1]. All manipulations of Mycobacterium tuberculosis prior to inactivation must be performed in a Class II Type A2 Biosafety Cabinet (BSC), as described in the protocol. Personal protective equipment (PPE) required for this protocol may include, but is not limited to: scrub clothing, bouffont cap, powered air-purifying respirator (PAPR) or N100 respirator, BSC gown, disposable gloves worn with double-gloving technique. Accel TB Disinfectant (0.5% hydrogen peroxide) is used for disinfection of materials prior to removal from a Class II Type A2 BSC or the removal of non-viable materials from the BSL-3 laboratory.
Biohazardous Waste Disposal: Accel TB Disinfectant is used for disinfection of biohazardous waste prior to decontamination by autoclaving.
Chemical Warnings: This protocol uses TRIzol reagent (containing guanidine thiocyanate and phenol) and chloroform. TRIzol is a chaotropic agent with serious physical and health hazards including for exposure routes airborne (inhalation), ingestion (swallowing), contact (skin, eyes, etc.). Likewise, chloroform is an organic solvent with serious health hazards for exposure routes airborne (inhalation), ingestion (swallowing), contact (skin, eyes, etc.). Refer to chemical safety data sheets (SDS) for detailed hazard information. Chemical reagents should be handled in a fumehood before use as specified in the protocol with the PPE described above.
Chemical Waste Disposal: Chemical waste including TRIzol and chloroform should not be mixed with bleach, exposed to high temperatures, nor autoclaved. Do not allow TRIzol reagent to come into contact with Accel TB Disinfectant, as they may react to form toxic gases. Chemical waste containers should be surface disinfected with 70% ethanol before disposal in accordance with local requirements.
Ethics statement
Not applicable.
Before start
In a biosafety level 3 (BSL-3) laboratory, prepare small-volume in vivo liquid cultures of Mycobacterium tuberculosis by inoculating BD BACTEC‱ MGIT‱ media prepared with BACTEC‱ MGIT‱ 960 Supplement and incubating the cultures at 37°C in an orbital shaker for 2-4 weeks. Estimate the exponential phase by comparing culture turbidity to a 0.9 McFarland Standard.
Total RNA Extraction from low-volume, in vitro Mycobacterium tuberculosis cultures
RNA is extracted from low-volume, in vitro cultures of Myocbacterium tuberculosis using variations on the manufacturer's protocol for the TRIzol Plus RNA Purification Kit and Phasemaker Tubes Complete System [2].
Place exponential phase liquid cultures of Mycobacterium tuberculosis On ice in a secondary container.
Note
During all further manipulations prior to TRIzol treatment,
keep samples on ice as much as possible.
Vortex the tubes containing liquid culture, then transfer contents (8 mL ) into a 15 mL conical tube.
Pellet the cells by centrifugation 4000 x g, 4°C, 00:20:00 . Remove and discard the supernatant from each conical tube, careful not to disturb the pellet.
20m
Resuspend the pellet in 1 mL water (mix by pipetting, do not vortex) and transfer resuspended cells to a 2 mL screw capped tube containing 0.1 mm zirconium beads.
Using a microcentrifuge, spin cells at 5.000 x g, 4°C, 00:10:00 . Remove and discard the supernatant, careful not to disturb the cell pellet or zirconium beads.
10m
Resuspend the pellet in 500 µL of autoclaved water (mix by pipetting, do not vortex). Using a microcentrifuge, spin cells at 5000 x g, 4°C, 00:10:00 . Discard the supernatant, careful not to disturb the pellet.
10m
Add 1 mL of TRIzol reagent to the pellet and gently vortex to homogenize.
Note
If the cell pellet is larger than approximately 100 µL, add more TRIzol reagent to maintain a 10:1 ratio to the cells.
Incubate the cells in TRIzol at Room temperature for 00:05:00 .
5m
Using a FastPrep-24 homogenizer or equivalent, bead beat for 3 cycles of 4 m/s for 1 minute each and rest on ice for 1 minute between cycles.
Optional safe stopping point, store at 4 °C overnight or -20 °C for up to a year.
Use microcentrifuge to spin the samples at 12.000 x g, 4°C, 00:05:00 to pellet cell debris.
5m
Transfer the supernatant (approximately 900-950 µL ) from the 2 mL tube to Phasemaker tube, careful not to touch the pellet or zirconium beads.
Incubate the samples for 00:05:00 at Room temperature to allow complete dissociation of the nucleoprotein complexes.
5m
Add 200 µL of chloroform to Phasemaker Tube (or proportional if >1 mL Trizol was added). Shake vigorously by hand for 00:00:30 to mix.
30s
Incubate the samples at Room temperature for 00:03:00 and then centrifuge the sample for 12.000 x g, 4°C, 00:05:00 .
8m
Transfer the aqueous phase to a new 2 mL flip cap tube (will be approximately 50% of the volume of the starting lysate).
Note
Phasemaker tubes contain an inert, thick liquid polymer, which forms a gel-like layer isolating the colourless upper aqueous phase containing RNA and the lower red phenol-chloroform phase following centrifugation. Be careful not to touch the Phasemaker Gel when removing the aqueous phase.
Add an equal volume (450-500 µL ) of 70% ethanol to the new tube containing aqueous phase, mix well by vortexing.
Transfer up to 700 µL of the sample to a spin cartridge with collection tube then centrifuge at 12.000 x g, Room temperature, 00:00:15 .
15s
Discard the column flow through, then re-insert the spin cartridge into the same collection tube.
Repeat Steps 18-19 until entire sample has processed.
Add 350 µL Wash Buffer I to the spin cartridge containing the bound RNA then centrifuge at 12.000 x g, Room temperature, 00:00:15 .
15s
Discard the flow through and collection tube, then insert the spin cartridge into a new collection tube.
Add 80 µL PureLink DNase mixture directly onto the surface of the spin cartridge membrane. Incubate at Room temperature for 00:15:00 .
15m
Add 350 µL Wash Buffer I to the spin cartridge and centrifuge at 12.000 x g, Room temperature, 00:00:15 .
15s
Discard the flow through and collection tube, insert the spin cartridge into new collection tube.
Add 500 µL of Wash Buffer II to the spin cartridge then centrifuge at 12000 rpm, Room temperature, 00:00:15 .
15s
Discard the flow through, then re-insert the spin cartridge into the same collection tube.
Repeat Steps 27-28 once more. .
Centrifuge the spin cartridge at 12.000 x g, Room temperature, 00:02:00 to dry the membrane.
2m
Discard the collection tube, then insert the spin cartridge into a labelled recovery tube.
Add 33 µL of RNase-free water to the center of the spin cartridge.
Incubate for 00:01:00 at Room temperature then centrifuge at 12.000 x g, Room temperature, 00:02:00 .
3m
Repeat Steps 32-33 twice more for a total of 3 sequential elutions. .
Discard the spin cartridge. The recovery tube contains the purified total RNA.
Store RNA extracts at -80 °C
Total RNA Quantification and Quality Check
Quantify the extracted RNA a Qubit 4.0 fluorometer with a RNA High Sensitivity Assay kit.
Note
The rRNA depletion and cDNA library preparation protocol described below requires 10 ng to 1 µg of total RNA for input.
Assess RNA purity using a NanoDrop‱ 2000 spectrophotometer.
Note
RNA samples with an A260/280 ratio near 2 are generally considered "pure" RNA. The expected A260/230 ratio for pure RNA is 2.0-2.2.
Citation
LINK
Determine RNA quality by RNA Integrity Number (RIN) determined by the Agilent 2100 Bioanalyzer System and RNA 6000 Nano kit.
Note
RIN is calculated by the Bioanalyzer System using an algorithm which considers the electrophoretic pattern of sample RNA, including the 16S:23S rRNA ratio, to determine the “intactness” of RNA on a scale of one to ten. The rRNA depletion and cDNA library preparation protocol described below requires that input total RNA has a RIN > 7.
Citation
LINK
Ribosomal RNA Depletion
Total RNA samples that met the requirements for integrity (RIN > 7) and quantity (10 ng – 1 µg) may proceed with rRNA depletion. Depletion of ribosomal RNA is performed using the manufacturer’s protocol for library preparation of intact RNA using NEBNext rRNA Depletion Kit (Bacteria) [3].
Dilute 10 ng - 1 µg of total RNA to a final volume of 11 µL in nuclease-free water, keeping On ice .
Note
Keep RNA samples on ice as much as possible before conversion to cDNA.
Add 2 µL NEBNext Bacterial rRNA Depletion Solution and 2 µL NEBNext Probe Hybridization Buffer to the 11 µL of total RNA On ice for a total volume of 15 µL .
Mix the reaction thoroughly by pipetting then briefly spin down the tube to collect the liquid from the sides of the tube.
Place the tube in a thermocycler and run the RNA/Probe Hybridization Reaction program below with the heated lid set to 105 °C :
Step 1: 95 °C for 00:02:00
Step 2: Ramp down to 22 °C at 0.1 °C per second
Step 3: Hold at 22 °C for 00:05:00
20m
Briefly spin down the tube and place On ice .
Add 2 µL of RNase H Reaction Buffer, 2 µL NEBNext Thermostable RNase H, and 1 µL Nuclease-free water to the 15 µL of hybridized RNA On ice for a total volume of 20 µL .
Mix the reaction thoroughly by pipetting then briefly spin down the tube to collect the liquid from the sides of the tube.
Incubate the RNase H digestion reaction in a thermocycler for 00:30:00 at 55 °C with the lid set to 55 °C .
30m
Briefly spin down the tube and place On ice .
To the 20 µL of RNase H-treated RNA, add 5 µL of DNase I Reaction Buffer, 2.5 µL of NEBNext DNase I, 22.5 µL and Nuclease-free water on ice for a total volume of 50 µL .
Mix the reaction thoroughly by pipetting then briefly spin down the tube to collect the liquid from the sides of the tube.
Incubate the DNase I digestion reaction in a thermocycler for 00:30:00 at 37 °C with the lid set to 40 °C (or off).
30m
Briefly spin down the tube and place On ice .
Purify the rRNA-depleted RNA using NEBNext RNA Sample Purification Beads in a 1.8X ratio: add 90 µL of NEBNext RNA Sample Purification Beads to the50 µL RNA suspension.
Note
Vortex the NEBNext RNA Sample Purification Beads well to resuspend before use.
Note
Agencourt RNAClean XP Beads may be used as an alternative to NEBNext RNA Sample Purification Beads.
Mix the RNA sample and beads thoroughly by pipetting and incubate On ice for 00:15:00 to allow the RNA to bind to the beads.
15m
Place the tubes on a magnetic plate for 00:05:00 to separate the beads from the supernatant.
5m
Remove and discard the clear supernatant (≤150 µL ) without touching the RNA-bound beads.
Add 200 µL of freshly prepared 80% ethanol to the RNA bound to the magnetic beads remaining in the tubes. Incubate for 00:00:30 at Room temperature then remove the supernatant.
30s
Repeat Step 58 once for a total of two washes.
Remove the tubes from the magnetic plate and air-dry for ≤ 00:05:00 at Room temperature to remove residual ethanol.
5m
Add 7 µL of nuclease-free water to each tube and mix thoroughly by pipetting.
Incubate the sample at Room temperature for 00:02:00 .
2m
Place the tubes on the magnetic plate for 00:02:00 to separate the magnetic beads from the RNA.
2m
Aspirate5 µL of the supernatant the containing rRNA-depleted RNA and transfer to a clean nuclease-free tube On ice .
Store rRNA-depleted RNA at -80 °C .
cDNA Library Preparation
15m
The cDNA library preparation from rRNA-depleted RNA is performed using the manufacturer's instructions for the NEBNext Ultra II Directional RNA Library Prep kit with Sample Purification Beads [3].
Add 4 µL of NEBNext First Strand Synthesis Reaction Buffer and 1 µL of Random Primers to the 5 µL of rRNA-depleted samples on ice for a total volume of 10 µL .
Fragment the RNA by placing the tubes on a thermocycler and incubating at 94 °C for 00:15:00 .
Note
Fragmentation conditions are optimized for generating 200 nucleotide inserts from intact RNA (RIN > 7).
15m
Immediately transfer the tube to ice.
Add 8 µL of NEBNext Strand Specificity Reagent and 2 µL of NEBNext First Strand Synthesis Enzyme Mix to the 10 µL of fragmented and primed RNA On ice for a total volume of 20 µL .
Keeping the solution on ice, mix the reaction thoroughly by pipetting.
Place the tube in a thermocycler with the heated lid set to 80 °C and run the following program for first strand cDNA synthesis:
Step 1: 00:10:00 at 25 °C
Step 2: 00:15:00 at 42 °C
Step 3: 00:15:00 at 70 °C
Step 4: Hold at 4 °C
40m
Add 8 µL of 10X NEBNext Second Strand Synthesis Reaction Buffer with dUTP Mix, 4 µL NEBNext Second Strand Synthesis Enzyme Mix, and 48 µL of Nuclease-free water to the 20 µL First-Strand Synthesis Product On ice for a total volume of 80 µL .
Keeping the solution on ice, mix the reaction thoroughly by pipetting.
Place the tube in a thermocycler for 01:00:00 at 16 °C with the heated lid set to 40 °C (or off) for second strand cDNA synthesis.
1h
Purify the double stranded cDNA with NEBNext Sample Purification Beads in a 1.8X ratio: add 144 µL of NEBNext Sample Purification Beads to the 80 µL cDNA suspension.
Note
SPRIselect Beads may be used as an alternative to NEBNext Sample Purification Beads.
Mix the cDNA sample and beads thoroughly by pipetting and incubate at Room temperature for 00:05:00 to allow the DNA to bind to the beads.
5m
Place the tubes on a magnetic plate for 00:05:00 to separate the beads from the supernatant.
5m
Remove and discard the clear supernatant (≤230 µL ) without touching the DNA-bound beads.
Add 200 µL of freshly prepared 80% ethanol to the RNA bound to the magnetic beads remaining in the tubes. Incubate for 00:00:30 at Room temperature then remove the supernatant.
30s
Repeat Step 80 for a total of two washes.
Remove the tubes from the magnetic plate and air-dry for ≤ 00:05:00 at Room temperature to remove residual ethanol.
5m
Add 53 µL of 0.1X TE Buffer to each tube and mix thoroughly by pipetting.
Incubate the sample at Room temperature for 00:02:00 .
2m
Place the tubes on the magnetic plate for 00:02:00 to separate the magnetic beads from the DNA.
2m
Aspirate50 µL of the supernatant the containing cDNA and transfer to a clean nuclease-free tube .
Store the purified double stranded cDNA at -20 °C .
Add 7 µL of NEBNext Ultra II End Prep Reaction Buffer and 3 µL of NEBNext Ultra II End Prep Enzyme Mix to the 50 µL of purified double stranded cDNA On ice for a total volume of 60 µL .
Mix the reaction thoroughly by pipetting then briefly spin down the tube to collect the liquid from the sides of the tube.
Place the tubes in a thermocycler with the heated lid set to 75C and run the following program for end-prep of the cDNA library:
Step 1: 00:30:00 at 20 °C
Step 2: 00:30:00 at 65 °C
Step 3: Hold at 4 °C
1h
Dilute the NEBNext Adaptor 5-fold in Adaptor Dilution Buffer before use (2.5 µL required per sample) and keep On ice .
Add 2.5 µL of diluted adaptor, 1 µL of NEBNext Ligation Enhancer, and 30 µL of NEBNext Ultra II Ligation Master Mix to the 60 µL of end prepped cDNA On ice for a total volume of 93.5 µL .
Mix the reaction thoroughly by pipetting then briefly spin down the tube to collect the liquid from the sides of the tube.
Place the tubes in a thermocycler and incubate at 20 °C with the heated lid off for 00:15:00
15m
Add 3 µL of USER Enzyme to the ligation mixture and mix thoroughly by pipetting. and incubate the tubes at 37*C Program 8B (Table 4) was run to complete the Ligation Reaction.
Place the tubes in a thermocycler at incubate at 37 °C with the heated lid set to 45 °C for 00:15:00 .
15m
Purify the ligation reaction with NEBNext Sample Purification Beads in a 0.9X ratio: add 87 µL of NEBNext Sample Purification Beads to the 96.5 µL DNA suspension.
Note
Vortex the NEBNext Sample Purification Beads well to resuspend before use.
Note
SPRIselect Beads may be used as an alternative to NEBNext Sample Purification Beads.
Mix the sample and beads thoroughly by pipetting and incubate at Room temperature for 00:10:00 to allow the DNA to bind to the beads.
10m
Place the tubes on a magnetic plate for 00:05:00 to separate the beads from the supernatant.
Remove and discard the clear supernatant (≤190 µL ) without touching the DNA-bound beads.
Add 200 µL of freshly prepared 80% ethanol to the RNA bound to the magnetic beads remaining in the tubes. Incubate for 00:00:30 at Room temperature then remove the supernatant.
Repeat Step 101 for a total of two washes.
Remove the tubes from the magnetic plate and air-dry for ≤ 00:05:00 at Room temperature to remove residual ethanol.
Add 17 µL of 0.1X TE Buffer to each tube and mix thoroughly by pipetting.
Incubate the sample at Room temperature for 00:02:00 .
Place the tubes on the magnetic plate for 00:02:00 to separate the magnetic beads from the DNA.
Aspirate15 µL of the supernatant the containing the ligated DNA and transfer to a clean nuclease-free tube .
Store adaptor-ligated cDNA samples at -20 °C
Perform PCR enrichment of adaptor ligated DNA with the unique dual index primer pairs supplied in the NEBNext Multiplex Oligos for Illumina kit: add 25 µL of NEBNext Ultra II Q5 Master Mix and 10 µL of Index Primer/i7 Primer Mix to the 15 uL of adaptor-ligated DNA (15 µL) On ice for a total volume of 50 µL .
Mix the reaction thoroughly by pipetting then briefly spin down the tube to collect the liquid from the sides of the tube.
Place the tube in a thermocycler with the heated lid set to 105C and run the following program for PCR enrichment of adaptor-ligated cDNA:
Step 1: 1 cycle
98 °C for 00:00:30
Step 2: 12 cycles
98 °C for 00:00:10
65 °C for 00:01:15
Step 3: 1 cycle
65 °C for 00:05:00
Step 4:
Hold at4 °C
Note
PCR-enrichment of cDNA is optimized based on ~100 ng of RNA input.
6m 55s
Purify the ligation reaction with NEBNext Sample Purification Beads in a 0.9X ratio: add 45 µL of NEBNext Sample Purification Beads to the 50 µL DNA suspension.
Note
Vortex the NEBNext Sample Purification Beads well to resuspend before use.
Note
SPRIselect Beads may be used as an alternative to NEBNext Sample Purification Beads.
Mix the sample and beads thoroughly by pipetting and incubate at Room temperature for 00:05:00 to allow the DNA to bind to the beads.
5m
Place the tubes on a magnetic plate for 00:05:00 to separate the beads from the supernatant.
Remove and discard the clear supernatant (≤100 µL ) without touching the DNA-bound beads.
Add 200 µL of freshly prepared 80% ethanol to the RNA bound to the magnetic beads remaining in the tubes. Incubate for 00:00:30 at Room temperature then remove the supernatant.
Repeat Step 116 for a total of two washes.
Remove the tubes from the magnetic plate and air-dry for ≤ 00:05:00 at Room temperature to remove residual ethanol.
Add 23 µL of 0.1X TE Buffer to each tube and mix thoroughly by pipetting.
Incubate the sample at Room temperature for 00:02:00 .
Place the tubes on the magnetic plate for 00:02:00 to separate the magnetic beads from the DNA.
Aspirate20 µL of the supernatant the containing the ligated DNA and transfer to a clean nuclease-free tube .
Store the cDNA libraries at -20 °C .
cDNA Library Pooling, Quantification & Quality Check
Quantify the cDNA libraries using a Qubit 4.0 fluorometer with a dsDNA High Sensitivity Assay kit.
Assess DNA purity using a NanoDrop‱ 2000 spectrophotometer.
Note
DNA samples with an A260/280 ratio near 1.8 are generally considered "pure" DNA. The expected A260/230 ratio for pure DNA is 2.0-2.2.
To achieve a minimum of 15 million paired-end reads per sample using a NextSeq 2000, pool 1 ng of cDNA from each of 20 samples to yield library concentrations greater than 750 picomolar (pM) .
Concentrate the pooled libraries using an Amicon Ultra-0.5 centrifugal filter unit according to the manufacturer's instructions:
Insert an Amicon Ultra 0.5 filter into the supplied microcentrifuge tube.
Transfer up to 500 µL of the library pool onto the filter and place the cap onto the filter/tube.
Note
Optional: if the volume is less than 500 µl, bring the volume up to 500 µl with nuclease-free water.
Place the filter/tube into a centrifuge, counter-balance, and spin at 14000 x g for 10 minutes.
If there is more than 500 µL of library pool to concentrate, discard the flow-through and repeat Steps 127.2 and 127.3 with the remaining volume in the same filter/tube.
Once the library pool has run through the filter (approximately 20 µL remaining), remove the filter from the existing microcentrifuge tube and place upside down into a clean microcentrifuge tube.
Place in a centrifuge with the cap open and cap facing inward into the centrifuge and spin at 1000 x g for two minutes to collect the filtered library pool.
Remove from the centrifuge and discard the filter. Retain the filtered library and cap the tube.
Size-select the pooled cDNA libraries at a range of 150-1000 bp using a BluePippin according to manufacturer's instructions.
Quantify the pooled cDNA libraries using a Qubit 4.0 fluorometer with a dsDNA High Sensitivity Assay kit.
Determine library size and quality using an Agilent 4200 TapeStation System with Genomic DNA ScreenTape Analysis reagents according to manufacturer's instructions.
RNA Sequencing
Sequence pooled cDNA libraries using the Illumina NextSeq 2000 platform with NextSeq 1000/2000 P2 (600 cycles) Reagents at a loading concentration of 700 picomolar (pM) according to manufacturer's instructions.
1d 18h
De-multiplex forward (R1) and reverse (R2) paired-end sequencing files according to sample indexing.
RNA-sequencing Data Pre-processing
The RNA-sequencing data pre-processing workflow is performed in Galaxy, an open-source environment for data analysis. The workflow is described in sequential steps below, and it may be downloaded in its entirety using the attached document: Galaxy-Workflow-Mycobacterial_PE-RNAseq_Data_Pre-processing.ga
Overview of the Mycobacterial PE-RNASeq Data Pre-Processing Workflow In Galaxy.
Citation
LINK
Assess pre-trimming quality of sequencing reads using FastQC (version 0.72).
Input: Paired-end reads as a dataset collection of paired R1/R2 FASTQ files
Output: Web FastQC Report for each FASTQ file
Expected result
- Expect >15 million reads per sample based on pooling and sequencing parameters.
- Expect a gyanosine/cytosine content (GC%) greater than 60% as M. tuberculosis has a GC-rich genome.
Example of FastQC basic statistics table showing > 15 million reads and 60% GC content before trimming and filtering.
Software
FastQC
NAME
Simon Andrews
DEVELOPER
Use Trimmomatic for adaptor trimming (ILLUMINACLIP), quality trimming
(SLIDINGWINDOW:4:20), and filtering out reads below 20 nt in length (MINLEN:20)
Input: (1) Paired-end reads as a dataset collection of paired R1/R2 FASTQ files; (2) Adaptor Sequences in FASTA format
Output: (1) Paired-end reads in which both reads in the pair survived filtering and trimming as a dataset collection of paired R1/R2 FASTQ files; (2) Reads in which one of the pair failed the filtering and trimming steps as a dataset collection of unpaired R1/R2 FASTQ files.
In future steps, move forward only with paired-end reads in which both pairs survived trimming.
Citation
LINK
Assess post-trimming read quality using FastQC (version 0.72).
Input: Paired-end reads in which both reads in the pair survived filtering and trimming as a dataset collection of paired R1/R2 FASTQ files
Output: Web FastQC Report for each FASTQ file
Expected result
- A threshold Phred score of 20 was used to ensure that high quality sequences are used for differential expression analysis.
- Desire >15 million reads to survive the trimming and quality filtering steps.
- Expect a gyanosine/cytosine content (GC%) greater than 60% as M. tuberculosis has a GC-rich genome.
- Expect high sequence duplication levels and overrepresented sequences, due to sequencing of highly expressed transcripts.
Example of FastQC per base sequence quality graph and Phred scores before trimming and filtering.
Example of FastQC per base sequence quality graph and Phred scores after trimming and filtering.
Example of FastQC basic statistics table showing >15 million reads and 60% GC content after trimming and filtering.
Example FastQC Sequence Duplication Level graph showing high levels of sequence duplication.
Use Kraken to detect potential contamination in three steps:
Citation
LINK
Use Kraken2 to assign taxonomic labels to each sequencing read. Use standard-ful Kraken database with prebuilt Refseq indexes including archaea, bacteria, viral, plasmid, human, UniVec_Core sequences
Input: Paired-end reads in which both reads in the pair survived filtering and trimming as a dataset collection of paired R1/R2 FASTQ files
Output: Collection of Kraken2 read classification files in tabular format
Run Kraken-report to obtain a summary of taxonomic classification for sequencing reads. This step determines the absolute number of reads assigned to each taxonomic level using the Kraken2 classification files generated in the previous step.
Input: Collection of Kraken2 read classification files in tabular format
Output: Collection of Kraken-report files in tabular format
Use Bracken to estimate the relative abundance of reads assigned at the genus level.
Input: Collection of Kraken-report files in tabular format
Output: Collection of tabular reports detailing number and fraction of reads assigned to given genera
Expected result
Example Bracken report showing >99% of reads assigned to Mycobacterium genus.
Align paired-end reads to the M. tuberculosis H37Rv reference genome (accession NC_000962.3) using Bowtie2.
Input: (1) Paired-end reads in which both reads in the pair survived filtering and trimming as a dataset collection of paired R1/R2 FASTQ files; (2) reference genome in FASTA format (accession NC_000962.3)
Output: Collection of alignments in BAM format
Citation
LINK
Determine alignment quality using Qualimap2 BamQC.
Input: Collection of alignments in BAM format
Output: Web QualiMap Report for each BAM alignment file
Expected result
- Expect >95% of reads to map to the reference genome.
- Expect duplication rate and overlapping read pairs to be high due to sequencing of highly expressed transcripts.
- Expect GC percentage to be >60%.
Example excerpt from QualiMap Report.
Citation
LINK
Use featureCounts (version 1.6.0.2) to determine read counts per gene using an annotated M. tuberculosis H37Rv reference genome (AL_123456.3).
Input: (1) Collection of alignments in BAM format; (2) annotated reference genome in GFF3 format (AL_123456.3)
Output: (1) featureCount tables listing the read count assigned to each geneid in tabular format; (2) featureCounts Summary showing the number of assigned and unassigned reads in tabular format.
Expected result
Example of first 14 lines of an Output featureCounts Table
Example of Output featureCounts Summary
Citation
LINK
Export/download the collection of featureCount tables in tabular format to be used as input for differential expression analysis.
Differential Expression Analysis
Normalization of read counts and differential expression analysis as described below is performed using DESeq2 (version 1.42.1), operating within R Studio. Before beginning, load the following libraries:
library(tidyverse)
library(janitor)
library(openxlsx)
library(DESeq2)
Citation
LINK
Create a metadata file in CSV format that includes the name of each featureCounts file downloaded in Step 141 as well as sample information that will be used for differential expression analysis.
| A | B | C | D | E | |
| id | strain | tx | pool | phenotype | |
| 1800686_1 | 1800686 | drug | 1 | R | |
| 1800686_2 | 1800686 | drug | 1 | R | |
| 1800686_3 | 1800686 | drug | 7 | R | |
| 1800686_7 | 1800686 | control | 5 | R | |
| 1800686_8 | 1800686 | control | 6 | R | |
| 1800686_9 | 1800686 | control | 6 | R | |
| 1800713_1 | 1800713 | drug | 1 | S | |
| 1800713_2 | 1800713 | drug | 1 | S | |
| 1800713_3 | 1800713 | drug | 2 | S | |
| 1800713_7 | 1800713 | control | 5 | S | |
| 1800713_8 | 1800713 | control | 6 | S | |
| 1800713_9 | 1800713 | control | 6 | S |
Example Metadata file in which columns show id (file name), strain (sample name), tx (treatment condition), pool (number of sequencing pool), and phenotype (sensitive or resistant).
Compile all featureCounts data into a single dataframe:
Create a log of files used for analysis called file_list:
Example Command:
file_list <- tibble(filename = list.files(path = "C:/Project_folder/", pattern = "*.tabular")) %>%
mutate(filepath = paste0("C:/Project_folder/", filename))
Note: all featureCount files must be in a single folder with the location specified in the command above.
Import all featureCount tables specified in file_list into a collection of dataframes called files:
Example Command:
files = lapply(file_list$filename, read.delim)
Compile the collection of featureCount dataframes into a single dataframe and tidy the data:
Example Commands:
raw_counts <- bind_cols(files, .name_repair = "universal") # bind dataframes
clean_counts <- dplyr::rename(raw_counts, "gene" = "Geneid...1") # rename column 1 to "gene"
clean_counts = clean_counts[,!grepl("*Geneid",names(clean_counts))] # remove redundant columns
clean_counts <- clean_names(clean_counts) # clean sample names
clean_counts <- rename_with(clean_counts, ~str_remove(.x, "x")) # clean sample names
Read metadata into a dataframe, convert all variables to factors, and set reference levels for each factor.
Example Commands:
metadata <- read.csv('metadata.csv', header = TRUE, sep = ",") # Import
metadata <- as.data.frame(unclass(metadata), stringsAsFactors = TRUE)
metadata[sapply(metadata, is.numeric)] <- lapply(metadata[sapply(metadata, is.numeric)], as.factor)
# Convert character and numeric variables to factors
metadata$tx <- relevel(metadata$tx, ref = "control")
metadata$phenotype <- relevel(metadata$phenotype, ref = "S")
# Set reference levels for each factor
Construct DESeqDataSet Object named dds from count data and metadata.
Example Command:
dds <- DESeqDataSetFromMatrix(countData=clean_counts,
colData=metadata,
design=~phenotype+ tx + phenotype:tx, tidy = TRUE)
Perform DESeq analysis on DESeqDataSet Object, generating large DESeqDataSet called analysis.
Example Commands:
analysis <- DESeq(dds,
test = "Wald",
fitType = "parametric",
sfType = "ratio")
resultsNames(analysis) # Returns the names of estimated effects of the model
Note
Consult the DESeq vignette to determine the most appropriate test, fitType, and sfType for your dataset.
Generate a results table (formal class DESeqResults) for each desired contrast.
Example commands:
# Example 1: Investigate effect of treatment (tx) on strains with a sensitive phenotype
result_table <- results(analysis,
contrast = c("tx", "control", "drug"),
alpha = 0.05)
# Example 2: Investigate the effect of treatment (tx) on strains with a resistant phenotype
result_table <- results(analysis, list(c("tx_drug_vs_control", "phenotypeR.txdrug")),
alpha = 0.05)
# Example 3: Investigate the difference between phenotypes with treatment
result_table <- results(analysis, list(c("phenotype_R_vs_S", "phenotypeR.txdrug")),
alpha = 0.05)
# Example 4: Investigate if the effect of treatment is different between phenotypes
interaction <- results(analysis, name = "phenotypeR.tx1drug",
alpha = 0.05)
Convert results tables from a formal class DESeqResults to a dataframe and export to an excel file.
Example Commands:
df_result <- as.data.frame(result_table) # convert formal class of DESeqResults into a dataframe
write.xlsx(df_result, file = "DESeq_results.xlsx", colNames = TRUE, rowNames = TRUE) # export as an excel file
Protocol references
Citation
LINK
Citation
LINK
Citation
LINK
Citations
PHAC. Biosafety Directive for Mycobacterium tuberculosis Complex (MTBC)
https://www.canada.ca/en/public-health/services/laboratory-biosafety-biosecurity/biosafety-directive-mycobacterium-tuberculosis-Invitrogen, ThermoFisher Scientific. User Guide: TRIzol Plus RNA Purification Kit and Phasemaker Tubes Complete System [Internet]
https://www.thermofisher.com/document-connect/document-connect.html?url=https://assets.thermofisher.com/TFS-Assets%2FLSG%2FmanuaNew England Biolabs. Protocol for Library Preparation of Intact RNA using NEBNext rRNA Depletion Kit (Bacteria) (NEB #E7850, NEB #E7860) and NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (NEB #E7760, NEB #E7765) [Internet]
https://www.neb.com/en/protocols/2019/09/18/protocol-for-library-preparation-of-intact-rna-using-nebnext-rrna-depletion-kit-bactStep 133
Galaxy Community. The Galaxy platform for accessible, reproducible, and collaborative data analyses: 2024 update.
https://doi.org/10.1093/nar/gkae410Step 135
Bolger AM, Lohse M, Usadel B. Genome analysis Trimmomatic: a flexible trimmer for Illumina sequence data.
https://doi.org/10.1093/bioinformatics/btu170Step 137
Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2.
https://doi.org/10.1186/s13059-019-1891-0Step 138
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2.
https://doi.org/10.1038/nmeth.1923Step 139
Okonechnikov K, Conesa A, García-Alcalde F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data.
https://doi.org/10.1093/bioinformatics/btv566Step 140
Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features.
https://doi.org/10.1093/bioinformatics/btt656Step 142
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2.
https://doi.org/Step 38
Glasel, J. Validity of nucleic acid purities monitored by 260nm/280nm absorbance ratios
PMID: 7702855.Step 39
Schroeder A, Mueller O, Stocker S, Salowsky R, Leiber M, Gassmann M, Lightfoot S, Menzel W, Granzow M, Ragg T. The RIN: an RNA integrity number for assigning integrity values to RNA measurements.
https://doi.org/