Dec 26, 2025
Rapid, large-scale and multi-species phylogenomic analysis using Covary
- Marvin De los Santos1
- 1ChordexBio



Protocol Citation: Marvin De los Santos 2025. Rapid, large-scale and multi-species phylogenomic analysis using Covary . protocols.io https://dx.doi.org/10.17504/protocols.io.261ge16rov47/v1
Manuscript citation:
https://doi.org/10.1101/2025.11.13.687960
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: December 24, 2025
Last Modified: December 26, 2025
Protocol Integer ID: 235778
Keywords: Rapid phylogenomics, large-scale phylogenomic analysis, multi-species phylogenomics, alignment-free phylogeny, machine learning phylogenetics, Covary, viral phylogenomics, SARS-CoV-2, Influenza A virus, Dengue virus, genome-scale sequence embedding, vector-based phylogeny, outgroup detection, species-level clustering, reproducible bioinformatics, Google Colab workflows, species phylogenomic analysis, comparative phylogenomic inference across diverse taxa, multiple viral species, free phylogenomic analysis, comparative phylogenomic inference, genomes of outbreak, scale analysis across multiple viral species, using complete genome sequence, genomic sequences into translation, complete genome sequence, encoding genomic sequence, genome, alphainfluenza virus, dengue virus, diverse taxa, outbreak, similarity inference, based similarity inference, measles virus, covary, aware vector representation, causing virus
Disclaimer
Your usage of different Covary versions, Covary-encoder, TIPs-VF and other components of the Covary suite may be limited. Please refer to the license notice at https://github.com/mahvin92/Covary?tab=License-1-ov-file for your review. If you implement Covary using a Colab notebook, please ensure to comply with Google’s Terms of Service. Note that this protocol was tested on Covary v2.1 using a computational performance on a free-tier subscription in Google Colab. Covary is provided "as is", without warranty of any kind, express or implied. For more information, please visit https://covary.chordexbio.com or read our paper at https://doi.org/10.1101/2025.11.13.687960.
Abstract
This protocol describes the use of Covary for fast, large-scale, multi-species phylogenomic analysis using complete genome sequences. The workflow is optimized for comparative phylogenomic inference across diverse taxa and is demonstrated using datasets associated with genomes of outbreak-causing viruses (SARS-CoV-2, dengue virus, measles virus, and alphainfluenza virus).
Covary enables alignment-free phylogenomic analysis by encoding genomic sequences into translation-aware vector representations and applying machine learning–based similarity inference. The protocol supports thousands-scale datasets, requires no coding experience, and is designed to run in a Google Colab environment without local software installation or maintenance.
This protocol focuses on phylogenomic-scale analysis across multiple viral species. Covary is accessible at https://covary.chordexbio.com/ or on GitHub at https://github.com/mahvin92/Covary.
Protocol Information
This protocol documents the application of Covary for: 1) multi-species phylogenomic comparison; 2) rapid inference of genome-scale relationships; 3) tree reconstruction across diverse taxa. The methods described here emphasize speed, scalability, and alignment-free analysis, making the workflow suitable for outbreak-scale genomics, cross-taxon comparisons, and exploratory phylogenomics.
The scope of this protocol is limited to tree inference and species-level classification using the complete genomes of SARS-CoV-2, dengue virus, measles virus, and alphainfluenza virus, as demonstrated previously (Rapid Phylogenomic Analysis of Thousands Outbreak‐Causing Viral Genomes Using Covary). This protocol is designed to be performed using Covary v2.1 on Google Colab.
Related Protocol
This protocol was modified from a previously described protocol "Machine learning-based phylogenetic analysis using Covary". The Covary version used and steps in performing an analysis were similar. Please read the related protocol for more information.
Data Acquisition
Download the genome sequences of SARS-CoV-2, dengue virus, measles virus, and alphainfluenza virus from The NCBI Viral Genomes Resource.
Visit the NCBI Virus portal at https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/
Search the name or taxid of the species below:
- SARS‐CoV‐2 (taxid: 2697049)
- Dengue virus (taxid: 12637)
- Alphainfluenza virus (taxid: 197911)
- Measles virus (taxid: 11234)
Refine the results by using the following filter parameters:
- Virus/Taxonomy = taxid
- Nucleotide Completeness= complete
- Ambiguous Characters= 0
Download 1,000 sequences for each group from the results.
Build a multi-FASTA file using the steps below:
- Click the "Download All Results" button
- Select "Nucleotide"
- Click "Next"
- Select "Download a randomized subset of all records (up to 2,000)"
- Select "Download a randomized subset of" and type "1000" in the input number field
- Click "Next"
- Select "Build custom" and obtain the: 1) Accession numbers and 2) Annotations of Pango lineage for SARS‐CoV‐2, the subtype for dengue virus, the genotype for alphainfluenza virus, and country of origin for measles virus.
- Click "Download"
Combine all the genome sequences from the four viral groups in one FASTA file (n=4,000). Note that .fasta is recommended but you can use a readable text formats like TXT/FNA/FA/etc.
Run quality and integrity check on your dataset using the recommended data preparation steps in the protocol for Machine learning-based phylogenetic analysis using Covary, Step 3. Preparing Your Data.
For the purpose of this exercise, you can download the compiled genome sequences at https://doi.org/10.6084/m9.figshare.30927302
Model Training
Pre-configure Covary to your specific requirements by modifying the parameters on Covary (Step 1. Set parameters). For guidelines, you may refer to the related protocol above (Pre-run Configuration step).
For the purpose of this exercise, change the value of the variable 'dlabel_fs' to 5 (or smaller) on Step 1.b. Plot customization on Covary. This will be helpful to resolve the data labels later on, since the default parameter (font size 12) is optimal for data max=500 entries (our training data is about 8 times more).
Step 1. Set parameters
a. Include sequence entries containing characters other than A, T, C, G (e.g., N)
include_N = "no"
b. Plot cutomization
[...]
dlabel_fs = 12 --> # change this to 5 or smaller
Run Covary by initiating or connecting to a runtime. Always initiate the run using the 'Run all' feature to make sure Covary is not interrupted, otherwise, execute the cells sequentially, one by one. Note: Always (if possible) connect to a GPU support (e.g., T4).
Toolbar: Click 'Runtime' --> Select 'Run all'
Upload your data in Step 2.
Code cell: Click 'Browse' --> Find your .fasta file --> Click 'Open' (or double click) --> Upload begins
Wait for Covary to finish learning from your data.
The results will be compiled and downloaded on Step 8. Note that Covary will run all the phylogenomic models, by default. Users with specific criteria can modify Step 7. Scoring and analysis for tree reconstruction, clustering, and embedding. To learn more about the phylogenomic models in Covary please read the related protocol above .
Run Validation
Validate the training results and parameters in Covary. Note that you can manually inspect the parameters used in training Covary with your data and assess the results on Step 6. Deep learning. The epoch count, the total number of batches, training duration, the loss and MAE, among others, are available for evaluation.
Inspect the pattern of clustering and pairwise distances of the embedding plots in PCA, t-SNE, and UMAP. Note that a well defined clustering pattern (either superimposition or closed grouping) in the different dimensionality reduction plots and presence of distinct blocks in the heatmaps are indicative of successful comparative sequence representation.
Figure 1. Genome‐scale embedding and similarity structure of the analyzed genomes from the four viral groups. A) Principal component analysis (PCA) of translation‐aware embeddings generated from the input data (4,000 complete genomes) of SARS‐CoV‐2, dengue virus, H5N* alphainfluenza virus, and measles virus. Each point represents a single viral genome inferred using Covary v2.1. Distinct clustering of genomes by viral classification is observed, indicating clear inter‐viral separation. B) Heatmap visualization of pairwise Euclidean distances across vector space for all analyzed viral genomes. Block‐like clustering patterns corresponding to the four viral groups are evident, reflecting high within‐group similarity and low between‐group similarity.
Assume or select the model that will suite your research objective. Note: You may refer to the 'Model Output Interpretation' and 'Manual Tree Search' in the related protocol for more information.
For the purpose of this exercise, PCA will be used to visualize the vector embeddings of the genomes. The PCA distance matrix will be used to analyze pairwise relationship of the sequences, and the complete hierarchal linkage was used to reconstruct the dendrogram.
Interpretation of Results
Inspect the quality and resolution of the generated dendrogram/s.
Evaluate the tree topologies, assessing clusters, clade formation, and branch length. Note that Covary is k-mer disparity-sensitive, and the branch length distance represents k-mer association profile rather than nucleotide composition. Thus, Covary v.2.1 does not represent evolutionary duration/time, yet.
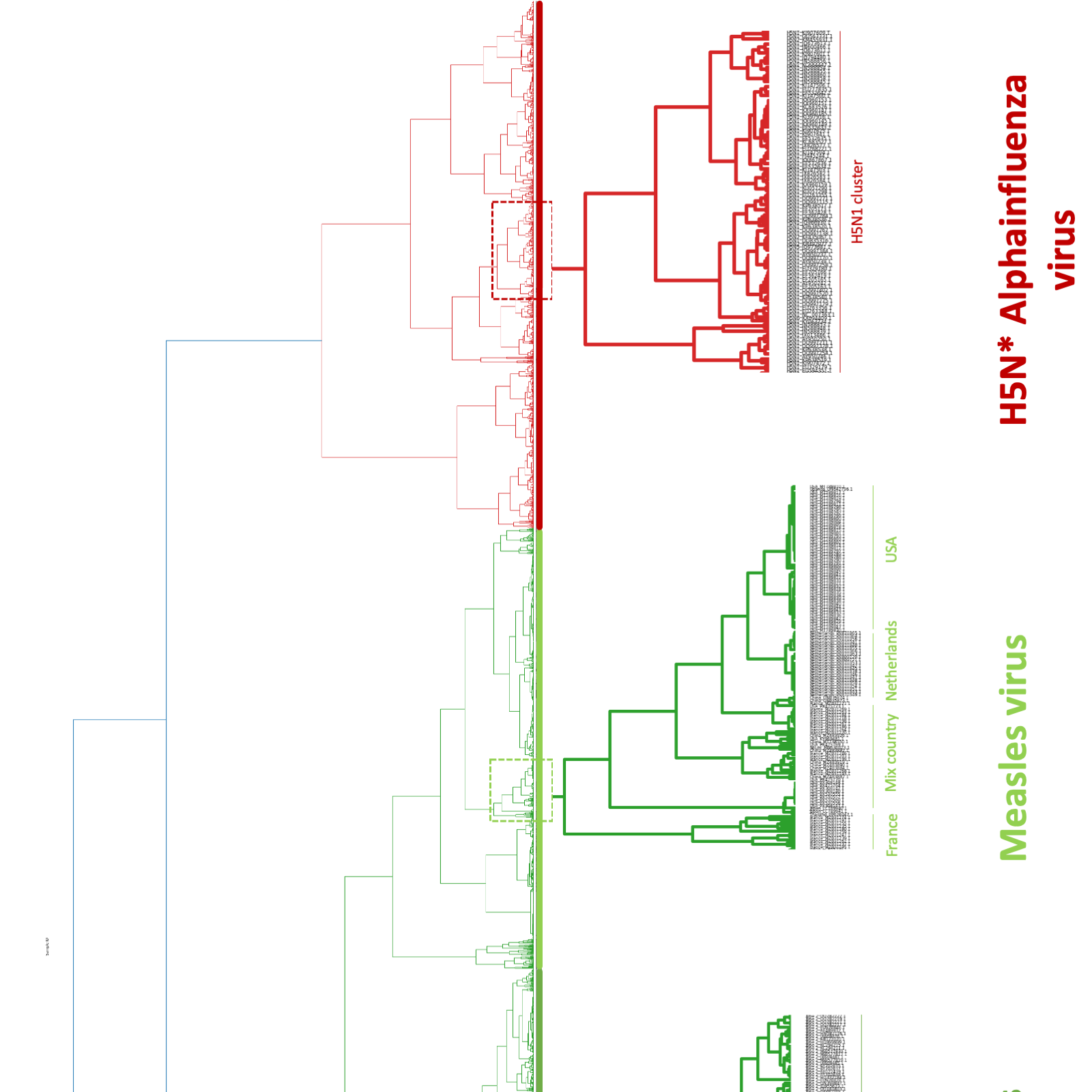
Figure 2. Hierarchical clustering reveals accurate taxonomic placing and ingroup classification. Dendrogram generated using complete linkage hierarchical clustering of the embeddings for the viral genomes used in this exercise. At higher hierarchical levels, genomes are grouped according to viral classification, forming four major clusters corresponding to SARS‐CoV‐2, dengue virus, alphainfluenza virus, and measles virus. At lower levels, ingroup structure is resolved, including SARS‐CoV‐2 Pango lineages, dengue virus subtypes, alphainfluenza virus clades, and measles virus groupings associated with geographic origin. These results illustrate the ability of Covary to capture both inter‐viral separation and biologically meaningful ingroup diversification using whole‐genome, alignment‐free representations.
Export the dendrogram into your preferred graphic annotation tool/software for labeling and data figure preparation.
Note that Covary results are interoperable with R and other python-based visualization libraries by importing and parsing the results files (in TSV format) into your workspace:
- Embeddings (dim1 & dim2)
- Pairwise Eucledian distance matrix
- Dendrogram linkage metrics
Protocol references
Covary: A translation-aware framework for alignment-free phylogenetics using machine learning. Cold Spring Harbor Laboratory. (2025). https://doi.org/10.1101/2025.11.13.687960
Rapid Phylogenomic Analysis of Thousands Outbreak‐Causing Viral Genomes Using Covary. MDPI AG. (2025). https://doi.org/10.20944/preprints202512.1970.v1
Machine learning-based phylogenetic analysis using Covary v2. Springer Science and Business Media LLC. (2025). https://doi.org/10.17504/protocols.io.n92ld13p8l5b/v2
TIPs-VF: An augmented vector-based representation for variable-length DNA fragments with sequence, length, and positional awareness. Cold Spring Harbor Laboratory. (2025). https://doi.org/10.1101/2025.02.15.637782