Sep 25, 2022
- 1University of California, Santa Cruz



Protocol Citation: Alison Tang 2022. R2C2 protocol draft. protocols.io https://dx.doi.org/10.17504/protocols.io.n2bvjx3knlk5/v1
Manuscript citation:
https://doi.org/10.1073/pnas.1806447115, https://doi.org/10.1101/2020.12.20.423532
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We have used this protocol with varying success with and without the size selection steps. It is developed by the Vollmers lab at UC Santa Cruz
Created: December 12, 2020
Last Modified: September 25, 2022
Protocol Integer ID: 45463
Keywords: cdna with the r2c2 protocol, genomic dna library preparation protocol, r2c2 cdna, resulting r2c2 cdna, r2c2 protocol, following oxford nanopore technology, oxford nanopore technology, nanopore, complementary dna, rna, total rna, selected cdna, cdna
Disclaimer
This is the R2C2 protocol developed by Dr. Roger Volden and Dr. Chris Vollmers, with help from other students in the Vollmers lab. I am consolidating the protocols developed by the Vollmers' lab into this protocol, adapted into protocols.io format. Any mistakes would be my (Alison Tang) own. This protocol is primarily being written for use in Dr. Angela Brooks' lab.
Abstract
Total RNA is reverse transcribed and PCR amplified using the smartseq2 system (with indexed oligo-dTs). Complementary DNA is then size selected for transcripts >=2.5 kb with a low melt agarose gel extraction. Size-selected cDNA and non-size-selected cDNA are pooled. Finally, long concatemers are generated from the cDNA with the R2C2 protocol. The resulting R2C2 cDNA can be sequenced on the nanopore following Oxford Nanopore Technology's genomic DNA library preparation protocol.
Image Attribution
Roger Volden
cDNA synthesis, adding oligo-dT indexes
1h 38m
Thaw oligo-dT, dNTP, DTT, Superase-In, 5x SmartScribe buffer, TSO SmartSeq primer on ice.
If you are size selecting, make four mixes of Mix #1 with four different oligo-dT indexes.
Otherwise, you will only need as many tubes of Mix #1 as you have samples that will be pooled together. So for 2 samples to be pooled together, you will need 2 tubes each with 2 ul of Mix #1 with two different oligo-dT indexes.
| A | B | |
| 1x, ul | ||
| oligo-dT, 10uM | 1 | |
| dNTP, 10mM | 1 |
Mix #1
Aliquot 2 µL from the tubes of mix #1 into each PCR tube (only necessary if you'll have multiple pools to be sequenced).
Make mix #2, 6 µL for each reaction.
| A | B | C | D | |
| 1x, ul | 4x, ul | 12.2x, ul | ||
| 5x SmartScribe Buffer | 2 | 8 | 24.4 | |
| DTT, 100mM | 1 | 4 | 12.2 | |
| TSO primer, 10uM | 0.3 | 1.2 | 3.66 | |
| H2O | 1.45 | 5.8 | 17.69 | |
| Superase-In | .25 | 1 | 3.05 | |
| SmartScribe RT (add last) | 1 | 4 | 12.2 |
Mix #2
Thaw the total RNA samples. Dilute each sample such that you have 4 µL if size selecting, or 2 µL if not, of 0.5 µg/µL total RNA.
Add 2 µL of diluted RNA to mix #1 with one oligo-dT index. If size selecting, then add the remaining 2 µL to another aliquot of mix #1 with a different oligo-dT index. Repeat for each sample.
Note
An example for two samples:
Oligo-dT index 1 - control sample, no size selection to be done
" " 2 - control sample, to be size-selected
" " 3 - experiment sample, no size selection
" " 4 - experiment sample, to be size-selected
Incubate tubes of Mix#1+RNA at 72 °C for 00:03:00 . Prepare to snap cool after.
3m
Snap cool on ice after denaturing to prevent secondary structure from reforming. Then pre-warm the thermocycler to 42 °C for RT reaction.
Add 6 µL mix #2 to each tube (10 µL total volume).
Incubate tubes of RNA+Mix#2 in thermocycler:
42 °C , 01:30:00
70 °C , 00:05:00
4 °C , 00:00:00 hold
1h 35m
cDNA amplification
29m 35s
Thaw ISPCR primers and KAPA Hifi HotStart on ice.
Make amplification mix (15 µL each reaction).
| A | B | C | |
| 1x, ul | 12.2x, ul | ||
| KAPA Hifi HotStart | 12.5 | 152.5 | |
| ISPCR primers, 10uM | 1 | 12.2 | |
| RNaseA | .75 | 9.15 | |
| Lambda Exonuclease | .75 | 9.15 |
Amplification mix
Add 15 µL amplification mix to each cDNA synthesis reaction (25 µL total volume).
Incubate tubes in thermocycler:
37 °C , 00:10:00
95 °C , 00:03:00
followed by 12 (or however many) cycles of:
98 °C , 00:00:20
67 °C , 00:00:15
72 °C , 00:06:30
then,
72 °C , 00:10:00 final extension
4 °C , 00:00:00 hold
30m 5s
Bead cleanup
Add 20 µL of Ampure XP beads (0.8:1 bead ratio) to enough low-bind tubes as you have PCR reactions.
Note
Wait for beads to come to room temperature. Vortex the beads gently before pipetting slowly.
Transfer the amplified product into the low bind tubes with beads. Pipette up and down 10 times, spin down, and then incubate at Room temperature for00:05:00 .
5m
Transfer tubes to a magnetic rack. Pipette off liquid. Wash the beads twice in 70% EtOH without disturbing the bead pellet.
Note
Use freshly made 70% EtOH. I leave the ethanol on for ~30 seconds each wash.
Elute in 20 µL of water. A smaller elution volume can be more effective for pooling the samples. Resuspend and then incubate for 00:10:00 at 37 °C . Then place on magnetic rack and remove eluate into new tubes.
10m
Qubit the purified cDNA (dsDNA HS).
Pool one control and one experimental sample together s.t. the concentration of the samples are equal. If you aren't size selecting, you can skip the size selection section. Else, pool the other ctrl and expt samples that contain the other two indexes. Make note of which two oligo-dT indexes will be size-selected.
Size selection (cDNA, gel extraction)
30m
Set up a 1% low melt agarose gel, ~100 ul TAE for a ~12 well gel with wide wells.
Note
We are interested in being able to sequence a 6 kb transcript. Thus, we size select to help enrich for these transcripts.
Melting this agarose can take a while. I was told to shake it every ~30 seconds in the microwave. One time I microwaved for ~20 minutes and a bit of agarose wouldn't melt. I ended up having to remove that piece of gel and adding TAE buffer back to make up the lost volume. Other times it all melted easily. Unclear why this is the case.
Load:
1 well -- diluted 1kb ladder, 10 ul, + 6X loading dye containing Sybr Gold
Other well(s) -- sample(s), ~30 ul, + 6X loading dye containing Sybr Gold
Run at 80V until separation around 2.5 kb is easier to see.
Transfer gel to blue light box. Excise 2.5 kb+ and place gel slice into a 2 ml tube.
Note
You should probably use a different blade for each sample if you have multiple.
Weigh gel slices, using an empty 2 ml tube to tare.
Note
If the weight of the gel slices exceeds ~600 g, the gel slices will need to be split into multiple tubes. This is because the slices + ~1,200 ul of buffer won't fit in a 2 ml tube.
Try to avoid this by making gel slices as small as possible and excising slivers of the gel that don't contain cDNA. If you end up splitting into multiple tubes, the final concentration of the sample will be much lower since they cannot be pooled effectively later.
Add β-agarase buffer to each tube. The μl of buffer to add is 2 volume's worth, or twice the number of grams of gel slice.
Incubate atOn ice for00:20:00 .
Note
NEB says 30 minutes.
20m
Remove buffer, then repeat steps 24 and 25.
Set up a heat block at 65 °C and another heat block at 42 °C .
Remove buffer, then incubate gel slices at 65 °C for00:10:00 .
10m
Transfer tubes to 42 °C heat block. Wait 1 minute for the melted gel to cool down to avoid inactivating beta-agarase in the next step.
Note
I use the cooldown time to measure the volume of melted gel using a P1000.
Add 2 ul of beta-agarase per 300 g of gel.
Note
NEB says 200 g.
Incubate at 42 °C for 01:00:00 , or overnight.
Note
One hour is from the protocol I was referring to when writing this, although in my limited experience 1 hour is not enough. I had multiple ~300 ul melted gels, added ~2 ul beta-agarase, incubated at 42C for ~70 minutes, and when I centrifuged I got a large (~half the volume) solid gel pellets. I had to re-melt and add more agarase. Overnight is definitely enough, I haven't had any residual gel from overnight incubations and I have done several of those.
1h
Place tubes On ice for 00:05:00 .
5m
Centrifuge at 15000 x g for 00:07:00 to pellet any undigested gel.
7m
Remove the supernatant and place into a low-bind tube.
Add 0.7:1 beads and follow the bead purification protocol above. Incubate sample and beads on a hula mixer. Elute in30 µL of water. Exact volume isn't too important here.
Note
You could even do a a higher bead ratio, funding permitting. There isn't anything to size-select out at this point, but you can't add too much as your magnet might not be strong enough for you to pipette liquid out w/o disturbing beads.
Qubit (dsDNA HS).
Combine size-selected ctrl+expt cDNA with non-size-selected ctrl+expt cDNA from step 18 s.t. the concentrations of all four oligo-dT indexes are comparable.
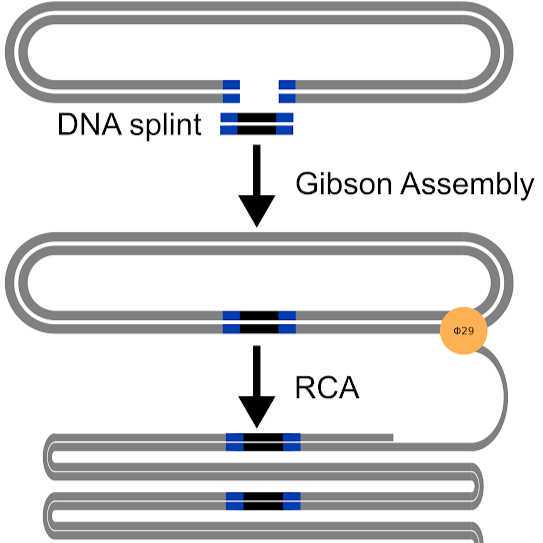
Circularization
8h
Combine 200 ng of splint with 200 ng of cDNA. Water can be used to adjust the volume to 20 µL if necessary.
| A | B | |
| ul | ||
| Pooled cDNA | x | |
| Splint | 10-x | |
| 2x NEBuilder Assembly Mix | 10 |
Note
If there is excess cDNA, you could increase the total volume from 20 µL to maybe 50 µL max. You could also do multiple splint assembly reactions. I've also seen 10 µL total volume splint assembly reactions, with then 40 ul phi29 mastermix instead of 30. I have also seen a 15 ul splint assembly reaction, +35 ul mastermix to get to 50 ul (referring to step 40). In this case, I noticed that the amount of exonuclease used was still 3 ul each, but the amount of NEB Buffer 2 was increased.
Also, you can get away with less splint if you need the volume for your cDNA.
Incubate at 50 °C for 01:00:00 .
1h
Prepare a mastermix of exonucleases, 30 µL per reaction.
| A | B | C | D | |
| 1x, ul | 3.2x, ul | 6.2x | ||
| NEB Buffer 2 | 5 | 16 | 31 | |
| H2O | 16 | 51.2 | 99.2 | |
| Exonuclease I | 3 | 9.6 | 18.6 | |
| Exonuclease III | 3 | 9.6 | 18.6 | |
| Lambda exonuclease | 3 | 9.6 | 18.6 |
Exonuclease mastermix
Add 30 ul of exonuclease mastermix to each reaction.
Incubate at 37 °C Overnight , or for at least 06:00:00 .
7h
Incubate at 80 °C for 00:20:00 to deactivate.
20m
Add40 µL of beads (0.8:1) to each reaction. Follow with bead cleanup protocol and elute in 30 µL H2O for three rolling circle amplification reactions, else elute in 10*(the number of RCA reactions desired).
Note
Three RCAs should be fine as long as you started with the recommended 200 ng of cDNA in the circularization step. I did four last time and it resulted in enough material to fit in 4-5 wells of a gel, so I was required to split each sample up into two gel extractions which isn't ideal.
Rolling circle amplification
8h
Make phi29 mastermix.
| A | B | C | |
| 1x, ul | 3.2x, ul | ||
| Phi29 buffer | 5 | 16 | |
| dNTP (10 mM) | 2.5 | 8 | |
| Random hexamer primers (exo resistant) | 2.5 | 8 | |
| H2O | 29 | 92.8 | |
| Phi29 polymerase | 1 | 3.2 |
phi29 mastermix
Add 10 µL of circularized cDNA to 40 µL of phi29 mastermix.
Incubate RCA reactions at 30 °C Overnight .
6h
Add 2 µL T7 endonuclease to each reaction. Pipette gently since the RCA cDNA is long.
Incubate for 02:00:00 at37 °C . Agitate occasionally.
Note
You can use this time to prepare the agarose gel (next section).
2h
Assemble a ~26 gauge needle with a syringe. Pull up the 3+ replicates of RCA T7 reactions into the syringe, combining them. Expel the reactions. Repeat the in+out four more times, for five times total. Three is ok too.
Clean the sheared combined RCA cDNA with the Zymo DNA Clean and Concentrator-5 kit. Use 2:1 binding buffer, change collection tubes to do an extra 1 minute dry spin, and elute in 20 µL . Exact volume isn't too important here.
Dilute samples 1:10 and qubit (dsDNA HS).
Size selection (RCA cDNA, gel extraction)
Set up a 1% low melt agarose gel, ~100 ul TE for a ~12 well gel with wide wells.
Load:
1+ wells -- diluted 1:50 NEB 1kb ladder, 10 ul, + 6X loading dye containing Sybr Gold
? wells -- sample + 6X loading dye containing Sybr Gold
Note
After DNA C&C-5, the RCA cDNA will be highly concentrated. Try to load <3 ug per well, so samples may need to be split into multiple wells. Keep in mind that more wells means more gel slices, which means you'll likely have to split the same cDNA sample into two tubes for the agarase cleanup, and only 1 ug of cDNA can go into the library prep anyway. I've found that anything more than 3 wells will need to be split.
Follow steps for running the gel, gel excision, buffer exchange, agarose digestion, bead cleanup (all detailed in the size selection section). You don't need to run the gel for as long since you only need to be able to cut out the 4+ kb molecules. Elute in 50 µL (nanopore SQK-LSK110 library prep takes 48 ul max, 1+ ug of cDNA).