Mar 24, 2026
Protocol for drug resistance screening for Plasmodium falciparum using Oxford Nanopore Sequencing
- Ajinkya Khilari1,
- Shweta Sharma2,
- Manali Bajpai3,
- Amit Sharma2,
- Dhanasekaran Shanmugam3
- 1CSIR-National Chemical Laboratory;
- 2International Centre for Genetic Engineering and Biotechnology;
- 3CSIR-NCL, Pune



External link: https://doi.org/10.1093/ofid/ofag106
Protocol Citation: Ajinkya Khilari, Shweta Sharma, Manali Bajpai, Amit Sharma, Dhanasekaran Shanmugam 2026. Protocol for drug resistance screening for Plasmodium falciparum using Oxford Nanopore Sequencing. protocols.io https://dx.doi.org/10.17504/protocols.io.x54v9233ml3e/v1
Manuscript citation:
Ajinkya Khilari, Shweta Sharma, Manali Bajpai, Anju Viswan K, Rini Chaturvedi, Bijay R Mirdha, Manju Rahi, Amit Sharma, Dhanasekaran Shanmugam, Targeted Genomic Surveillance Unveils Genetic Variations Linked to Regional Malaria Drug Resistance Dynamics in India, Open Forum Infectious Diseases, Volume 13, Issue 3, March 2026, ofag106, https://doi.org/10.1093/ofid/ofag106
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: July 13, 2024
Last Modified: March 24, 2026
Protocol Integer ID: 103370
Keywords: sequencing antimalarial drug resistance, antimalarial drug resistance in plasmodium falciparum necessitate, antimalarial drug resistance, plasmodium, protocol for drug resistance screening, plasmodium falciparum necessitate, robust molecular surveillance tool, based malaria treatment, malaria treatment, drug resistance screening, resistance gene, detection at parasitemia level, using oxford nanopore, sequencing, sequencing approach, samples with high human dna background, oxford nanopore, novel resistance, parasitemia level, targeted multiplex pcr, associated mutation, clinical sample, primer pool
Abstract
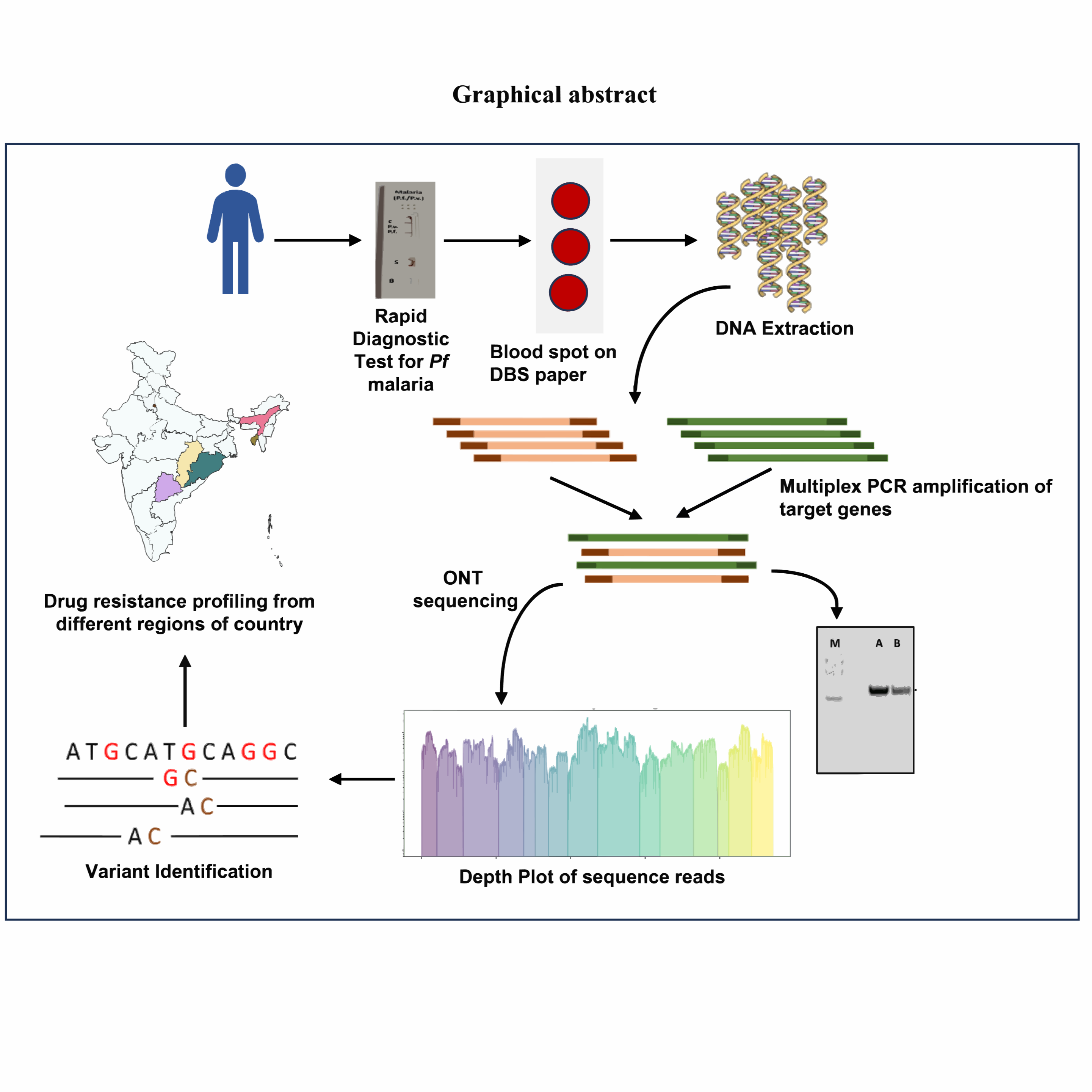
Antimalarial drug resistance in Plasmodium falciparum necessitates robust molecular surveillance tools. This protocol describes a targeted multiplex PCR–based amplicon sequencing approach (PfMDR15 panel) for genotyping 15 key drug-resistance genes using Oxford Nanopore sequencing. The method employs two primer pools to generate ~1.5 kb amplicons from clinical samples, including dried blood spots. It demonstrates high sensitivity, enabling detection at parasitemia levels as low as 0.01%, and performs reliably even in samples with high human DNA background. Sequencing is followed by alignment and variant calling to identify known and novel resistance-associated mutations. The workflow is rapid, scalable, and cost-effective, making it suitable for routine surveillance. This protocol provides a practical framework for monitoring regional resistance patterns and supports evidence-based malaria treatment and control strategies.Antimalarial drug resistance in Plasmodium falciparum necessitates robust molecular surveillance tools. This protocol describes a targeted multiplex PCR–based amplicon sequencing approach (PfMDR15 panel) for genotyping 15 key drug-resistance genes using Oxford Nanopore sequencing. The method employs two primer pools to generate ~1.5 kb amplicons from clinical samples, including dried blood spots. It demonstrates high sensitivity, enabling detection at parasitemia levels as low as 0.01%, and performs reliably even in samples with high human DNA background. Sequencing is followed by alignment and variant calling to identify known and novel resistance-associated mutations. The workflow is rapid, scalable, and cost-effective, making it suitable for routine surveillance. This protocol provides a practical framework for monitoring regional resistance patterns and supports evidence-based malaria treatment and control strategies.
Guidelines
1. Scope of the Protocol
This protocol describes a targeted multiplex PCR-based amplicon sequencing workflow for genotyping 15 Plasmodium falciparum drug-resistance genes using Oxford Nanopore sequencing. The method is optimized for clinical samples (e.g., dried blood spots) and enables detection of resistance-associated mutations across geographically diverse populations .
2. Ethical and Regulatory Compliance
- Ensure all sample collection procedures are approved by an Institutional Ethics Committee.
- Obtain informed consent from all participants prior to sample collection.
- Follow national and institutional guidelines for handling human biological samples.
- Ensure compliance with biosafety level requirements applicable to malaria samples.
3. Laboratory Safety Guidelines
- Wear appropriate PPE (lab coat, gloves, eye protection) at all times.
- Treat all biological samples as potentially infectious, even after DNA extraction.
- Do not remove samples or reagents from the laboratory.
- Dispose of biological and chemical waste according to institutional biosafety rules.
- Handle mutagenic agents (e.g., ethidium bromide) with caution.
4. Sample Requirements
Sample type:
- Dried Blood Spots (DBS) or whole blood
Inclusion criteria:
- Confirmed P. falciparum infection (e.g., RDT or 18S PCR)
Storage conditions:
- Transport at 4°C
- Long-term storage at −80°C
5. DNA Quality Requirements
DNA should be:
- High-quality and minimally degraded
- A260/280 ratio ≥ 1.8
Minimum input DNA:
- ≥ 50 ng for ONT sequencing
- Low parasitemia samples (as low as 0.01%) are supported by this protocol
6. Principle of the Method
This protocol uses:
- Multiplex PCR amplification of 15 resistance-associated genes
- Two primer pools (Pool A and Pool B) to avoid interference
- Amplicon size ~1.5 kb for optimal Nanopore sequencing
- Sequencing using ONT platforms followed by variant analysis
- This approach enables high sensitivity genotyping even in samples with high human DNA background .
7. Target Genes Included (PfMDR15 Panel)
- The protocol targets 15 key genes associated with antimalarial resistance, including:
- pfcrt (chloroquine resistance)
- pfmdr1 (multidrug resistance)
- pfdhfr, pfdhps (SP resistance)
- pfk13 (artemisinin resistance)
- pfaat1 (emerging lumefantrine resistance marker)
- Additional genes: pfmrp1, pfmdr2, pfatpase6, pfdhodh, pfcytb, etc.
8. Critical Considerations
- Avoid Cross-Contamination
Use separate areas for:
- DNA extraction
- PCR setup
- Post-PCR handling
- Change gloves between samples
Primer Handling
- Keep primer pools separate (Pool A vs Pool B)
- Thaw on ice and mix gently
PCR Optimization
- Ensure balanced amplification across all targets
- Validate amplification using gel electrophoresis
Bead Purification
- Do not over-dry beads
- Use fresh 80% ethanol
ONT Flow Cell Handling
- Avoid introducing air bubbles
- Use gentle pipetting
- Check pore availability before loading
9. Citation
If you use this protocol, please cite:
Ajinkya Khilari, Shweta Sharma, Manali Bajpai, Anju Viswan K, Rini Chaturvedi, Bijay R Mirdha, Manju Rahi, Amit Sharma, Dhanasekaran Shanmugam, Targeted Genomic Surveillance Unveils Genetic Variations Linked to Regional Malaria Drug Resistance Dynamics in India, Open Forum Infectious Diseases, Volume 13, Issue 3, March 2026, ofag106, https://doi.org/10.1093/ofid/ofag106
Protocol materials
Ethidium bromide 10 mg/mlMerck MilliporeSigma (Sigma-Aldrich)Catalog #E1510
1X TAE Buffer
Invitrogen™ UltraPure™ AgaroseInvitrogen - Thermo FisherCatalog #16500100
GeneRuler 100 bp Plus DNA Ladder, ready-to-useThermo Fisher ScientificCatalog #SM0323
AMPure XPBechman CoulterCatalog #A63882
Rapid Barcoding Kit 96 V14 (SQK- RBK114.96)Oxford Nanopore TechnologiesCatalog #SQK- RBK114.96
RepliQa HiFi ToughMix® VWR International (Avantor)Catalog #95200-500
Safety warnings
This protocol involves handling biological samples, PCR reagents, and sequencing equipment. Although it does not require high-risk pathogens or highly hazardous chemicals, strict adherence to laboratory safety and good experimental practices is essential to ensure user safety, sample integrity, and reliable results.
1. Biological Safety
• Clinical samples (e.g., blood or dried blood spots) must be treated as potentially infectious.
• Perform all handling using appropriate biosafety practices.
• Always wear personal protective equipment (PPE) including gloves, lab coat, and mask where necessary.
• Avoid direct contact with biological material and prevent aerosol generation.
2. Contamination Risk
• This protocol involves high-sensitivity PCR, making it highly susceptible to contamination.
• Use separate work areas for:
1. DNA extraction
2. PCR setup
3. Post-PCR processing
4. Always change gloves between samples.
5. Use filtered pipette tips and sterile consumables.
6. Cross-contamination can lead to false-positive variant detection.
3. Sample Handling and Storage
• Improper storage or temperature fluctuations can result in DNA degradation, affecting downstream analysis.
• Maintain:
1. 4°C during transport
2. −80°C for long-term storage
3. Avoid repeated freeze–thaw cycles of DNA samples.
4. DNA Quality and Quantification
• Poor-quality DNA (e.g., A260/280 < 1.8) may lead to:
• Failed PCR amplification
• Uneven sequencing coverage
• Inaccurate genotyping
• Always verify DNA quality before proceeding.
5. Primer and PCR Handling
• Avoid mixing or cross-contaminating primer pools (Pool A and Pool B).
• Improper handling can result in:
1. Non-specific amplification
2. Loss of multiplex balance
3. Thaw primers on ice and mix gently before use.
6. Magnetic Bead Purification
• Over-drying beads can reduce DNA recovery.
• Rough pipetting can cause DNA shearing, impacting sequencing quality.
• Ensure proper ethanol removal before elution to avoid downstream inhibition.
7. Oxford Nanopore Flow Cell Handling
• Flow cells are highly sensitive and expensive components.
• Avoid:
1. Introducing air bubbles during priming or loading
2. Applying excessive pipetting force
3. Improper handling may lead to:
• Loss of active pores
• Reduced sequencing output
8. Chemical Safety
• Reagents used in gel electrophoresis (e.g., ethidium bromide or alternatives) may be toxic and mutagenic.
• Handle with care and dispose of waste according to institutional guidelines.
9. Instrument Usage
• Follow manufacturer instructions strictly for all instruments (e.g., thermal cycler, Qubit, Nanopore devices).
• Incorrect usage may lead to:
1. Equipment damage
2. Data loss or poor sequencing quality
10. Data Integrity
• Mislabeling samples or inconsistent tracking can compromise the entire dataset.
• Maintain accurate and consistent sample IDs throughout the workflow.
11. Interpretation of Results
• Detection of mutations does not always directly translate to phenotypic drug resistance.
• Interpret results using validated databases and published literature.
12. General Laboratory Conduct
• Do not eat, drink, or store food in the laboratory.
• Use a dedicated notebook for recording experimental observations.
• Wash hands thoroughly after completing work.
Ethics statement
Experiments involving animals must be conducted according to internationally-accepted standards and should always have prior approval from an Institutional Animal Care and Use Committee (IACUC) or equivalent ethics committee(s). Ethics approval should be obtained before performing these experiments. If approval was obtained, please include the name of the IACUC or equivalent ethics committee and any relevant permit numbers.
Sample Collection
20m
Please follow the protocol described in the link below for blood sample collection on DBS paper
DNA Extraction from DBS
1d
Please follow the link provided below for DNA extraction from DBS
Selective Multiplex Gene Amplification using Plasmodium falciparum Multi Drug Resistance 15 panel (Pf MDR15)
6h 30m
PCR amplification of Drug resistance genes for P. falciparum from field samples is carried out for genotyping. The PCR amplification of 15 genes is done in multiplexed manner.
- The PCR primers were designed using the reference genome PlasmoDB68_Pfalciparum3D7 for Plasmodium falciparum available in PlasmoDB using PrimalScheme
- A total of 37 primer pairs were designed to cover all the 15 drug resistant genes and each PCR product is ~1.5kb in sizewith ~0.5kb overlap between adjacent PCR fragments.
- The Pf MDR15 primer set is divided into pool A and pool B such that adjacent amplicons are in different pools. The details of the entire primer set is given in Table 01
| A | B | C | D | E | F | G | |
| Primer | Pool | Primer Sequence | Primer Size | GC content | Tm | Gene | |
| PfMDR15_1 | A | AGCCATTTTTGTATTCCCAAATAGCT | 26 | 34.62 | 60.29 | DHFR | |
| PfMDR15_2 | A | ATTCAGCACCGAAATGTCTCCA | 22 | 45.45 | 60.47 | DHFR | |
| PfMDR15_3 | B | GCAATAGGATAAATGTTATATTGTCTAGAACC | 32 | 31.25 | 59.94 | DHFR | |
| PfMDR15_4 | B | CCGTTCAGGTAATTTTGTCATCATTTG | 27 | 37.04 | 60.42 | DHFR | |
| PfMDR15_5 | A | TCCGTTAATAATAAATACACGCAGTCA | 27 | 33.33 | 59.78 | CRT | |
| PfMDR15_6 | A | TCCCTTGTCATGTTTGAAAAGCA | 23 | 39.13 | 59.55 | CRT | |
| PfMDR15_7 | B | TGTTGTAACAATAGCTCTTGTAGAAATGA | 29 | 31.03 | 60.22 | CRT | |
| PfMDR15_8 | B | TGGTTCTCTTACAACATCACCCT | 23 | 43.48 | 59.61 | CRT | |
| PfMDR15_9 | A | TCTTGGGAAGAAACACAGTCGT | 22 | 45.45 | 60.01 | CRT | |
| PfMDR15_10 | A | AGAGATCTCTATACCTTCAACATTATTCCT | 30 | 33.33 | 60.25 | CRT | |
| PfMDR15_11 | B | CGTGAAAATTCTATTGTAATACTATGGTCAATG | 33 | 30.3 | 60.89 | CYTB | |
| PfMDR15_12 | B | TATAGTTTTTGGCGGCTGAGCA | 22 | 45.45 | 60.8 | CYTB | |
| PfMDR15_13 | A | CCAATATATTTGGTATGCGTATTACATGAA | 30 | 30 | 59.57 | MRP1 | |
| PfMDR15_14 | A | AGAATATAACCACTTCAACTATATCAGAGGA | 31 | 32.26 | 60.52 | MRP1 | |
| PfMDR15_15 | B | TCGAGATAAAAGAATTGATAACATGCATCA | 30 | 30 | 60.88 | MRP1 | |
| PfMDR15_16 | B | GGAAGGATCTAAAGATGTAAATATATCATCAAG | 33 | 30.3 | 59.66 | MRP1 | |
| PfMDR15_17 | A | TGCCAATCTTATATGTTCCTCAAAATAGT | 29 | 31.03 | 60.12 | MRP1 | |
| PfMDR15_18 | A | TCTTACTTGCTTTTGTAATATTGTGTCTGA | 30 | 30 | 60.69 | MRP1 | |
| PfMDR15_19 | B | ACTCATCCAGTTTTAAGGGTTCCA | 24 | 41.67 | 60.22 | MRP1 | |
| PfMDR15_20 | B | AGCATGAACAATTTCATCATCTGTGA | 26 | 34.62 | 60.29 | MRP1 | |
| PfMDR15_21 | A | AGGCTGTATTTATCATGTCATACTCCT | 27 | 37.04 | 60.26 | MRP1 | |
| PfMDR15_22 | A | TGTATGTATTATGTATGCATGGGTGTG | 27 | 37.04 | 60.1 | MRP1 | |
| PfMDR15_23 | B | GGCGTAAATATTCGTGTTATAATTTCTCC | 29 | 34.48 | 60.12 | K13 | |
| PfMDR15_24 | B | TCTACACCATCAAATCCACCTATACA | 26 | 38.46 | 59.83 | K13 | |
| PfMDR15_25 | A | GGAAACGATTTGATGAAGAAAGATTAAGA | 29 | 31.03 | 59.67 | K13 | |
| PfMDR15_26 | A | GGGAAAATCATAAACAATCAAGTAATGTGT | 30 | 30 | 60.34 | K13 | |
| PfMDR15_27 | B | CGTTGAACTTATTATATCTTTGTCATTCGT | 30 | 30 | 59.86 | ATP6 | |
| PfMDR15_28 | B | TCCTCTTAGCACCACTCCTACT | 22 | 50 | 59.87 | ATP6 | |
| PfMDR15_29 | A | CGGATGATGGAGAAGAAGGATCA | 23 | 47.83 | 59.93 | ATP6 | |
| PfMDR15_30 | A | CAGCTGATTTCAATGCTGGTGC | 22 | 50 | 61.17 | ATP6 | |
| PfMDR15_31 | B | GGTGATAATATTAATACGGCCAGAGC | 26 | 42.31 | 60.18 | ATP6 | |
| PfMDR15_32 | B | TTAATTTTCTTGGTTCTTTGCTCTTCC | 27 | 33.33 | 59.56 | ATP6 | |
| PfMDR15_33 | A | CAAGCCGAGACACAAAAACGAC | 22 | 50 | 61.02 | ATP6 | |
| PfMDR15_34 | A | GTGGCACCTTTTTATAACGTATCGT | 25 | 40 | 60.31 | ATP6 | |
| PfMDR15_35 | A | ATTCTGTTCATTTTGGTAAATACTAACAG | 29 | 28 | 54 | TCTP | |
| PfMDR15_36 | A | CGTCAAAGTTTGTTAAAATGTGTTTAATAAATGG | 34 | 26 | 57 | TCTP | |
| PfMDR15_37 | B | ATGTTCAACAAGATCCATTTGAAGTACCAG | 30 | 37 | 60 | TCTP | |
| PfMDR15_38 | B | ACCCATTTGATGTTTATTAAATGAAAGGG | 29 | 31 | 57 | TCTP | |
| PfMDR15_39 | A | ATCGATACCAAGTATTAAATGAACACCT | 28 | 32.14 | 59.61 | DHPS | |
| PfMDR15_40 | A | TCATCTTCTGATCCACACAATCACA | 25 | 40 | 60.67 | DHPS | |
| PfMDR15_41 | B | TTTGTAAATGATCCTCTTAGTATGTTGGT | 29 | 31.03 | 59.81 | DHPS | |
| PfMDR15_42 | B | AGTTGATCCTTGTCTTTCCTCATGT | 25 | 40 | 60.61 | DHPS | |
| PfMDR15_43 | A | TGGTCCTTTTGTTATACCTAATCCAAAA | 28 | 32.14 | 59.87 | DHPS | |
| PfMDR15_44 | A | TCATTTGAACCAGTCAGGAAATGAA | 25 | 36 | 59.55 | DHPS | |
| PfMDR15_45 | B | TGTGTGATAGATAGCTCCAGTCGA | 24 | 45.83 | 61.01 | DHODH | |
| PfMDR15_46 | B | AGTTTTGCTCCGCTAACACCTC | 22 | 50 | 61.31 | DHODH | |
| PfMDR15_47 | A | TGGTGGAAAAATATTAAGTAATGATAGGCA | 30 | 30 | 60.4 | DHODH | |
| PfMDR15_48 | A | CACTTATGTGTCGCCCGTG | 19 | 58 | 57 | DHODH | |
| PfMDR15_49 | B | GTGTACATAGCTTATTTCATTTATAAGATTTAG | 33 | 24 | 54 | MDR1 | |
| PfMDR15_50 | B | ACACATCAACAACATCAGAATCTTTAATAG | 30 | 30 | 59.76 | MDR1 | |
| PfMDR15_51 | A | TTGGAGTTGTTAGTCAAGATCCATTATT | 28 | 32.14 | 59.71 | MDR1 | |
| PfMDR15_52 | A | AATGCAAAAACTCCGCTTGACA | 22 | 40.91 | 60.08 | MDR1 | |
| PfMDR15_53 | B | TGCTCTTTCTGGTTAGCATGGT | 22 | 45.45 | 60.14 | MDR1 | |
| PfMDR15_54 | B | CAATATAACGGACAAGAGTTGATACTGTTC | 30 | 37 | 58 | MDR1 | |
| PfMDR15_55 | A | GATATATGAAAATTGTGAAAACAAAATTGTGTG | 33 | 24 | 56 | MDR2 | |
| PfMDR15_56 | A | ATTAATCCTTCTATTGTTGCCGGAAT | 26 | 34.62 | 59.67 | MDR2 | |
| PfMDR15_57 | B | TCACAAGTACAACAATCAGCTTTTATAGA | 29 | 31.03 | 60.22 | MDR2 | |
| PfMDR15_58 | B | ATGTGGTTCATTTGATTTTGATTGCA | 26 | 30.77 | 59.73 | MDR2 | |
| PfMDR15_59 | A | AGCAAATGAGATGGATAATGTATATCATGA | 30 | 30 | 59.95 | MDR2 | |
| PfMDR15_60 | A | TATCAAAAGTGCTTTTAATATTTGCCGAAG | 30 | 30 | 57 | MDR2 | |
| PfMDR15_61 | B | GCTTTATAAGGTGGAAAATAATAAAGTTGAAC | 32 | 28 | 56 | Fd | |
| PfMDR15_62 | B | GTGCATTATATTCAAAAATATTCTGGAGTACC | 32 | 31 | 57 | Fd | |
| PfMDR15_63 | A | CGAAAAGAGTATATTTTCCTTTTTCCTTCA | 30 | 30 | 59.91 | EXO | |
| PfMDR15_64 | A | TCGTTATCGTCATCGTAATCCTTAACA | 27 | 37.04 | 60.95 | EXO | |
| PfMDR15_65 | B | ACGAATGGAGTCATTTAGCAGCA | 23 | 43.48 | 60.87 | EXO | |
| PfMDR15_66 | B | AGGGGATAAGGTTTATTTTGTAGGGA | 26 | 38.46 | 60 | EXO | |
| PfMDR15_67 | A | ACCTGAAGACGTTAAAAATGTAAAGTACA | 29 | 31.03 | 60.57 | EXO | |
| PfMDR15_68 | A | AATTATACGATTTGGAGGGCGTAAAT | 26 | 34.62 | 59.73 | EXO | |
| PfMDR15_69 | B | GCACACTCTTCTTCTTGATTTTCCTTATCG | 30 | 40 | 59.8 | ARPS10 | |
| PfMDR15_70 | B | CCACATGGGGAGATCGCAAAAAG | 23 | 52 | 60 | ARPS10 | |
| PfMDR15_71 | A | TCCTCAGTTTATTGTAGCAGGCCC | 24 | 50 | 60.3 | ARPS10 | |
| PfMDR15_72 | A | CGTGAAATACATAGAAAAATTAAGAACAATGCC | 33 | 30 | 57.9 | ARPS10 | |
| PfMDR15_73 | B | AGAAACAAAGCATGAACAGAGTT | 23 | 35 | 54.9 | AAT | |
| PfMDR15_74 | B | ACAAGAAGAACACAACTACTGGT | 23 | 39 | 55.8 | AAT |
Table 01 - List of primer squences for PfMDR15 panel.
- For individual primers 100µmolar stocks are prepared in Nuclease Free Water and stored in -20 °C
- For multiplexed genomic PCR reaction the pool A and pool B are prepared by separately mixing 10 µL of each of the pool A primers in one pool and 10 µL of each of the pool B primers in the other pool. The primer pools can be stored in -20 °C
- At the time of setting up the PCR reactions the pool A and pool B primer pools are diluted 10 times m(10µmolar primer mix) with Nuclease Free Water and added to the reactions as given in Table 02
# NOTE: Primer pools can be ordered directly from the manufacturer as ready-to-use 10µmolar primer mix.
- Pool A and Pool B PCR reactions are set up separately using RepliQa HiFi ToughMix® VWR International (Avantor)Catalog #95200-500 master mix
| A | B | |
| repliQa HiFi ToughMix® | 12.5 μL | |
| Primer Mix of Pool A or Pool B | 3.75 μL | |
| Template DNA | 5-50 ng | |
| Nuclease Free Water | Makeup volume to 25 μL |
Table 02 - PCR reaction mix composition using PfMDR15
- PCR amplification conditions for genomic PCR reaction is given in Table 03
| A | B | C | D | |
| Step | Temperature | Time | Cycles | |
| Initial Denaturation | 95 ⁰C | 1 min | X 1 cycle | |
| Denaturation | 95 ⁰C | 15 sec | X 35 cycles | |
| Annealing and Extension | 63 ⁰C | 5 min | ||
| Hold | 4 ⁰C | ∞ |
Table 03 – PCR conditions for multiplex whole genome amplification using LSDV_WGSPP_3.5 panel
Precautions
- Make sure to avoid cross-mixing of primers between the pool A and pool B sets
- Primer mix and DNA samples must be thawed on ice
- Gently mix primer pools (vortexing) and DNA samples (flicking) and spin down before use
- After every use store primer mix and DNA samples in -20 °C to prevent degradation
6h
Analysis of PCR products by agarose gel electrophoresis
# NOTE: This is an optional step and the PCR products can be directly sequenced without analysis by agarose gel electrophoresis. However, this step is recommended for two reasons- 1) to confirm that the pool A and pool B PCRs have worked for each sample before sequencing; 2) to ensure that the PCR product is devoid of primer dimers (this can affect sequence data quality).
- Reagents required are -
Invitrogen™ UltraPure™ AgaroseInvitrogen - Thermo FisherCatalog #16500100
GeneRuler 100 bp Plus DNA Ladder, ready-to-useThermo Fisher ScientificCatalog #SM0323
Ethidium bromide 10 mg/mlMerck MilliporeSigma (Sigma-Aldrich)Catalog #E1510
1X TAE Buffer
- 5 µL of PCR product is loaded on the agarose gel and electrophoresis is carried out at 120mV for 00:30:00
- A 3.5kb band should be seen in pool A and pool B reactions of each sample. Only samples for which both pool A and pool B reactions have worked can be taken forward for sequencing
30m
Sample Purification for PCR Products (optional)
40m
# NOTE: This is an optional step but is highly recommended if primer dimers are detected in the PCR product
- Add AMPure XPBechman CoulterCatalog #A63882 bead slurry to each sample in 1:1 volumetric ratio
- Incubate at Room temperature for 00:15:00 to allow DNA binding to beads
- Keep the sample tubes on magnetic stand for separation of beads bound to DNA
- Carefully remove supernatant without disturbing the beads
- Wash twice with 150 µL freshly prepared 80% Ethanol without disturbing the beads
- Keep for drying in Room temperature for 00:00:30 to 00:01:00 to remove excess ethanol. NOTE: Ensure that the beads do not dry completely.
- Remove tube from magnetic stand and add 25 µL of Nuclease Free Water (NFW). Mix the beads properly by pipetting
- Incubate for 00:15:00 at Room temperature to separate DNA from the beads
- Keep the tube on magnetic stand and wait for beads to separate out
- Recover the eluate (~20µl) containing DNA into a fresh tube
Precautions
- Always mix the beads only by pipetting or flicking to avoid DNA shear
- Always use freshly prepared 80% ethanol for washing
- Avoid carryover of beads while recovering the eluate in the last step
40m
Oxford Nanopore Sequencing Steps Using SQK- RBK114.96 Kit
2h 5m
# NOTE: In the publication citing this protocol older reagents and R9.4 .1 flowcells were used. But since these reagents are discontinued by ONT, protocol mentioned below is updated with respect to new reagents compatible with R10.4.1 flowcells.
- If bead purification (Step No. 13) was omitted, go directly to barcoding step
- If bead purification of PCR product done, do QC using nanodrop and check ratios i.e. A260/280 and A260/230
- For the samples with good quality of DNA (as given in Step No. 5.2), Qubit readings are taken to determine DNA concentration .
- Atleast 50ng of DNA per sample is required for sequencing
10m
Barcoding
- Between 50 ng to 100 ng of each purified PCR product is taken in maximum volume of 9 µL (make up volume with NFW if needed) and 1 µL sequencing rapid barcode (RB01-96) provided in the kit is added
# NOTE: If bead purification was not done, 9 µL of PCR product can be used directly in this step
- Mix properly by pipetting atleast 10 times
- Incubate at following conditions-
30 °C for 00:02:00 , then at 80 °C for 00:02:00 and then at 4 °C or On ice for 00:05:00
- Pool all the barcoded samples together in a single 1.5 mL microfuge tube
- All further steps are carried out on this pooled barcoded sample
20m
Purification of pooled barcoded sample
- Add AMPure XPBechman CoulterCatalog #A63882 bead slurry to each sample in 1:1 volumetric ratio
- Incubate at Room temperature for 00:15:00 to allow DNA binding to beads
- Keep the sample tubes on magnetic stand for separation of beads bound to DNA
- Carefully remove supernatant without disturbing the beads
- Wash twice with 1 mL freshly prepared 70% Ethanol without disturbing the beads
- Keep for drying in Room temperature for 00:00:30 to 00:01:00 to remove excess ethanol. NOTE: Ensure that the beads do not dry completely.
- Remove tube from magnetic stand and add 20 µL of Elution Buffer (EB) provided in the kit. Mix the beads properly by pipetting
- Incubate for 00:15:00 at Room temperature to separate DNA from the beads
- Keep the tube on magnetic stand and wait for beads to separate out
- Recover the eluate (~15µl) containing barcoded library into a fresh tube
Precautions
- Always mix the beads only by pipetting or flicking to avoid DNA shear
- Always use freshly prepared 70% ethanol for washing
- Avoid carryover of beads while recovering the eluate in the last step
# NOTE: At this stage the barcoded library can be stored at 4 °C for 1 day and at -20 °C for longer duration (upto 1 month)
30m
Adapter ligation of barcoded library
- Take 11 µL of barcoded library and add 1 µL of diluted rapid adapter (1.5 µL Rapid Adapter (RA) + 3.5 µL Adapted Buffer (ADB))
- Incubate at 37 °C for 00:10:00 and immediately proceed with loading the library on the flowcell and sequencing
#NOTE: The sequencing library prepared is compatible with version 10 flowcells (FLOMIN_114) from Oxford Nanopore Technology (refer to manufacturer's website for latest update on flowcells and compatible kits).
10m
Flow cell priming
# NOTE: This step can be performed during incubation period for adapter ligation (Step No. 14.3)
- Make priming mix by adding 30 µL Flow Cell Tether (FCT) in 1170 µL Flow Cell Flush (FCF) and mix by vortexing
- Remove waste from waste port if required (# NOTE: Make sure that the spot-on port and priming port are CLOSED during this step)
- Set 200 µL reading in 1000 µL pipette then OPEN priming port and suck air out by bringing pipette volume to 220 µL - 230 µL to avoid entry of air bubble while loading (# NOTE: Make sure that the spot-on port is CLOSED during this step)
- Add 800 µL of priming mix via the priming port. It is important to follow the method given here for this step. (# NOTE: Make sure that the spot-on port is CLOSED during this step)
- Incubate at room temperature for 00:05:00 to 00:10:00
- Do second priming of flow cell by adding 200 µL priming mix. It is important to follow the method given here for this step. (# NOTE: Make sure that the spot-on port is OPENED during this step)
Precautions
- While following priming steps, strictly avoid air bubble entry in the flowcell as this will result in pore loss
- Follow pipetting steps with care and check when to open or close spot-on port and priming port as per recommendation
15m
Sample preparation for loading
- Add 37.5 µL Sequencing Buffer (SB) and 25.5 µL Library Beads (LIB) to adapter ligated sequencing library
- OPEN spot-on port and add the prepared sample to spot-on port using 100 µL pipette. NOTE: Strictly avoid air bubble entry into spot-on port
- CLOSE both spot-on and priming port and remove any solution from waste port
Precautions
- While loading the sample, strictly avoid air bubble entry in the flowcell as it will result in pore loss
- Avoid leaving any liquid in the waste port after loading as this might block the ports and prevent reuse of flowcell if needed
10m
Starting the sequencing
- Open MinKNOW software
- Click on the left panel and select Start
- Then select start sequencing from the menu
- Select the flow cell position and enter your experiment name and sample ID
- Then click on kit selection and select-Rapid Barcoding Kit 96 V14 (SQK- RBK114.96)Oxford Nanopore TechnologiesCatalog #SQK- RBK114.96
- Move forward to select parameters as required and start the sequencing
10m
Oxford Nanopore Sequencing Steps Using SQK- NBD114.96 Kit
2h 9m 15s
# NOTE: This section mentions the alternate barcoding protocol for library preparation. The protocol should be used if required more data since native barcoding kit generates three times more data compared to rapid barcoding kit.
- Perform bead purification of PCR product, do QC using nanodrop and check ratios i.e. A260/280 and A260/230
- For the samples with good quality of DNA (as given in Step No. 5.2), Qubit readings are taken to determine DNA concentration .
- Atleast 200ng of DNA per sample is required for sequencing.
Thaw the DNA Control Sample (DCS) at room temperature, mix by vortexing, and place on ice.
Prepare the NEBNext Ultra II End Repair / dA-tailing Module reagents in accordance with manufacturer's instructions, and place on ice:
For optimal performance, NEB recommend the following:
- Thaw all reagents on ice.
- Ensure the reagents are well mixed. Note: Do not vortex the Ultra II End Prep Enzyme Mix.
- Always spin down tubes before opening for the first time each day.
- The NEBNext Ultra II End Prep Reaction Buffer may contain a white precipitate. If this occurs, allow the mixture to come to room temperature and pipette the buffer several times to break up the precipitate, followed by a quick vortex to mix.
End prep
- In a clean 96-well plate, aliquot 200 fmol (130 ng for 1 kb amplicons) of DNA per sample.
- Make up each sample to 11.5 µL using nuclease-free water. Mix gently by pipetting and spin down.
- Combine the following components per well:
Between each addition, pipette mix 10-20 times.
| Reagents | Volume | |
| 200 fmol amplicon DNA | 11.5 µl | |
| Diluted DNA Control Sample (DCS) | 1 µl | |
| Ultra II End-prep Reaction Buffer | 1.75 µl | |
| Ultra II End-prep Enzyme Mix | 0.75 µl | |
| Total | 15 µl |
Table 4- End prep reaction for per sample
- Ensure the components are thoroughly mixed by pipetting and spin down briefly.
- Using a thermal cycler, incubate at 20 °C for 00:10:00 and65 °C for 00:10:00 .
#Note Take forward the end-prepped DNA into the native barcode ligation step.
If users want to pause the library preparation here, we recommend cleaning up your sample with 1X AMPure XP Beads (AXP) and eluting in nuclease-free water before storing at 4 °C .
20m
00:20:00 Barcoding Ligation
- Prepare the NEB Blunt/TA Ligase Master Mix according to the manufacturer's instructions, and place on ice: 1. Thaw the reagents at room temperature. 2. Spin downthe reagent tubes for 00:00:05 3. Ensure the reagents are fully mixed by performing 10 full volume pipette mixes.
- Thaw the AMPure XP Beads (AXP) at room temperature and mix by vortexing. Keep the beads at room temperature.
- Thaw the EDTA at room temperature and mix by vortexing. Then spin down and place on ice.
- Thaw the Short Fragment Buffer (SFB) at room temperature and mix by vortexing. Place on ice.
- Thaw the Native Barcodes (NB01-96) required for your number of samples at room temperature. Individually mix the barcodes by pipetting, spin down, and place them on ice.
- Select a unique barcode for every sample to be run together on the same flow cell. Up to 96 samples can be barcoded and combined in one experiment.
- In a new 96-well plate, add the reagents in the following order per well mixing well by pipetting between each addition:
| Reagent | Volume | |
| Nuclease-free water | 3 µl | |
| End-prepped DNA | 0.75 µl | |
| Native Barcode (NB01-96) | 1.25 µl | |
| Blunt/TA Ligase Master Mix | 5 µl | |
| Total | 10 µl |
Table 5- Barcoding ligation reaction per sample
- Thoroughly mix the reaction by gently pipetting and briefly spinning down.
- Incubate for00:20:00 at room temperature.
- Add 2 µl EDTA (blue cap) to each well and mix thoroughly by pipetting and spin down briefly.
- Pool the barcoded samples in a 1.5 mL Eppendorf DNA LoBind tube.
- Resuspend the AMPure XP Beads (AXP) by vortexing.
- Add 0.4 % volume AMPure XP Beads (AXP) to the pooled reaction, and mix by pipetting.
- Incubate on a Hula mixer (rotator mixer) for00:10:00 at room temperature.
- Spin down the sample and pellet on a magnet for 00:05:00 Keep the plate on the magnetic rack until the eluate is clear and colourless, and pipette off the supernatant.
- Wash the beads with700 µL of Short Fragment Buffer (SFB). Flick the beads to resuspend, spin down , then return the sample to the magnetic rack and allow the beads to pellet. Remove the buffer using a pipette and discard.
- Repeat the previous step.
- Spin down and place the tube back on the magnetic rack. Pipette off any residual buffer.
- Remove the tube from the magnetic rack and resuspend the pellet in 35 µL nuclease-free water by gently flicking.
- Incubate for 00:10:00 at 37 °C Every 00:02:00 agitate the sample by gently flicking for 10 seconds to encourage DNA elution.
- Pellet the beads on a magnetic rack until the eluate is clear and colourless.
- Remove and retain 35 µL of eluate into a clean 1.5 mL Eppendorf DNA LoBind tube.
1h 7m 5s
Adapter ligation and clean-up
- .Prepare the NEBNext Quick Ligation Reaction Module according to the manufacturer's instructions, and place on ice.
- Spin down the Native Adapter (NA) and Quick T4 DNA Ligase, pipette mix and place on ice.
- Thaw the Elution Buffer (EB) at room temperature and mix by vortexing. Then spin down and place on ice.
- Thaw either Long Fragment Buffer (LFB) or Short Fragment Buffer (SFB) at room temperature and mix by vortexing. Then spin down and keep at room temperature.
- In a 1.5 mL Eppendorf LoBind tube, mix in the following order: Between each addition, pipette mix 10-20 times.
| Reagent | Volume | |
| Pooled barcoded sample | 30 µl | |
| Native Adapter (NA) | 5 µl | |
| NEBNext Quick Ligation Reaction Buffer (5X) | 10 µl | |
| Quick T4 DNA Ligase | 5 µl | |
| Total | 50 µl |
Table 6 - Adapter Ligation Reaction
- Thoroughly mix the reaction by gently pipetting and briefly spinning down.
- Incubate the reaction for 00:20:00 at room temperature.
- Resuspend the AMPure XP Beads (AXP) by vortexing.
- Add 20 µL of resuspended AMPure XP Beads (AXP) to the reaction and mix by pipetting.
- Incubate on a Hula mixer (rotator mixer) for00:10:00 at room temperature.
- Spin down the sample and pellet on the magnetic rack. Keep the tube on the magnet and pipette off the supernatant.
- Wash the beads by adding either125 µL Long Fragment Buffer (LFB) or Short Fragment Buffer (SFB). Flick the beads to resuspend, spin down, then return the tube to the magnetic rack and allow the beads to pellet. Remove the supernatant using a pipette and discard.
- Repeat the previous step.
- Spin down and place the tube back on the magnet. Pipette off any residual supernatant.
- Remove the tube from the magnetic rack and resuspend pellet in 15 µL Elution Buffer (EB).
- Spin down and incubate for 00:10:00 at 37 °C Every 00:02:00 , agitate the sample by gently flicking for 00:00:10 to encourage DNA elution.
- Pellet the beads on a magnet until the eluate is clear and colourless, for at least 1 minute.
- Remove and retain15 µL of eluate containing the DNA library into a clean 1.5 ml Eppendorf DNA LoBind tube.
#Note For further steps follow sub steps 6.4, 6.5 and 6.6
42m 10s
Data Analysis
- In-house bash script named as AmpVarPro was developed for automated data analysis, accessible from github via https://github.com/ajinkyakhilari/AmpVarPro.git
- All steps involved in data analysis and the corresponding programs are described in https://github.com/ajinkyakhilari/AmpVarPro.git
20m
Protocol references
1. Oyola SO, Ariani CV, Hamilton WL, et al. Whole genome sequencing of Plasmodium falciparum from dried blood spots using selective whole genome amplification. Malar J 2016; 15:597.
2. Girgis ST, Adika E, Nenyewodey FE, et al. Drug resistance and vaccine target surveillance of Plasmodium falciparum using nanopore sequencing in Ghana. Nat Microbiol 2023; 8:2365–77.
3. de Cesare M, Mwenda M, Jeffreys AE, et al. Flexible and cost-effective genomic surveillance of P. falciparum malaria with targeted nanopore sequencing. Nat Commun 2024; 15:1413.
4. MalariaGEN. DBS01: Dried Blood Spot (DBS) Collection Protocol. Version 1.2. Oxford: MalariaGEN; 2021.
5. Musale P, Khilari A, Gade R, et al. Identification of genetic variations linked to buparvaquone resistance in Theileria annulata infecting dairy cattle in India. PLoS One 2025; 20:e0326243.
6. Bajpai M, Khilari A, Likhitkar B, et al. Detection and variant characterization of lumpy skin disease virus from dairy cattle in India. Virus Evol 2025; 11:veaf090.
7. Quick J, Grubaugh ND, Pullan ST, et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc 2017; 12:1261–76.