Aug 07, 2024
Phylogenetic Read Placement Protocol
- Matthew Hays1,
- Clara Fuchsman1
- 1University of Maryland Center for Environmental Science



Protocol Citation: Matthew Hays, Clara Fuchsman 2024. Phylogenetic Read Placement Protocol . protocols.io https://dx.doi.org/10.17504/protocols.io.4r3l2q24xl1y/v1
Manuscript citation:
Fuchsman, C. A., Garcia Prieto, D., Hays, M. D., & Cram, J. A. (2023).
Associations between picocyanobacterial ecotypes and cyanophage host genes
across ocean basins and depth. PeerJ, 11, e14924.
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: May 22, 2024
Last Modified: August 07, 2024
Protocol Integer ID: 100319
Keywords: Phylogenetic read placement , Metagenome, Bioinformatics, phylogenetic read placement of metagenomic read, phylogenetic read placement protocol, phylogenetic read placement, metagenomic read, phylogenetic tree, metagenomic sample, creation of phylogenetic tree, assembly of metagenome, metagenome, taxonomic identification, collation by taxonomy, proteins from assembled contig, raxml, tree
Abstract
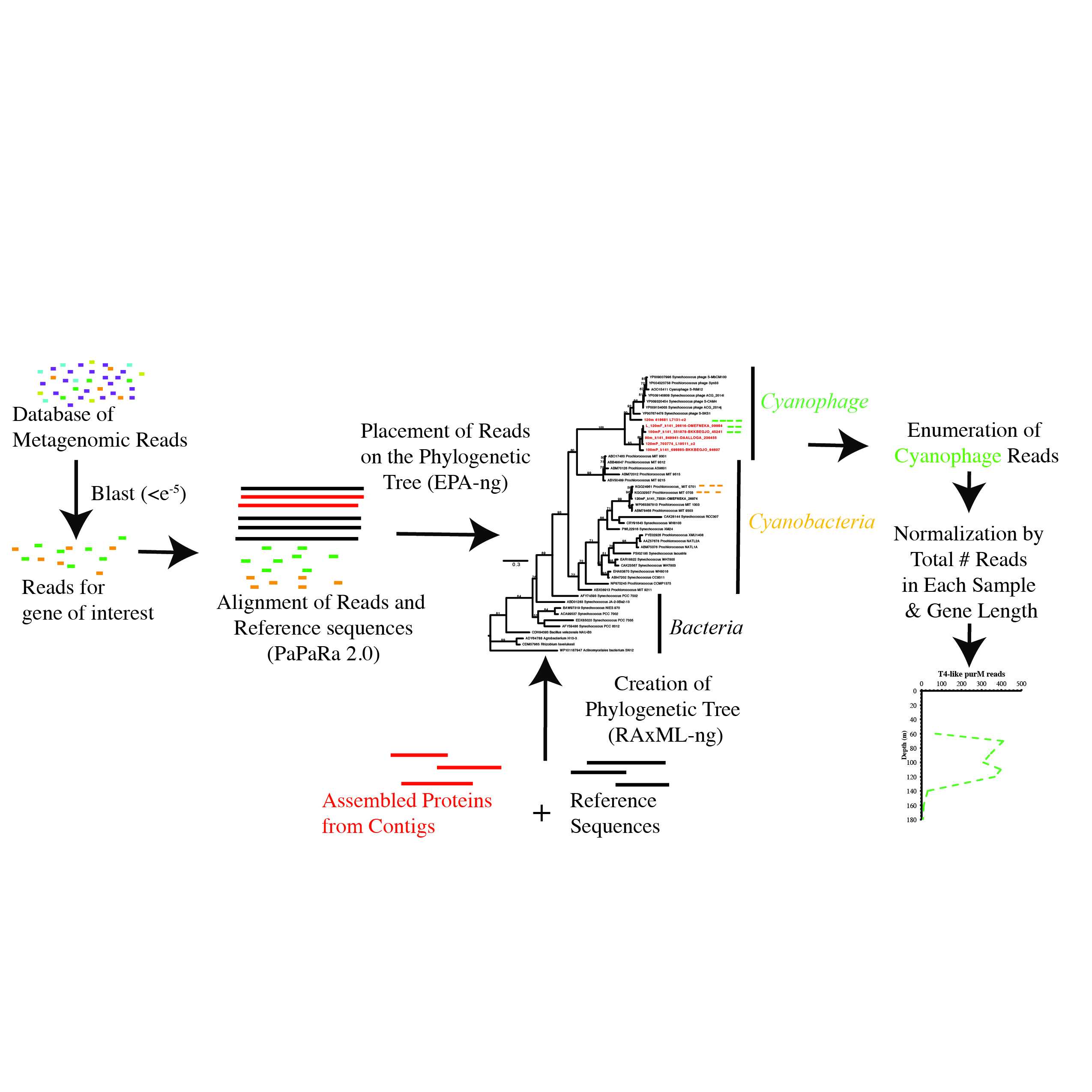
This protocol is for phylogenetic read placement of metagenomic reads in as quantitative a manner as possible without addition of known standards. This protocol includes assembly of metagenomes into contigs, creation of phylogenetic trees including proteins from assembled contigs, the phylogenetic read placement of metagenomic reads onto the tree, taxonomic identification, and collation by taxonomy and metagenomic sample. Several published software are used in this protocol, but here we share our scripts for using the outputs from this software in as quantitative a manner as is possible. This protocol is a daughter protocol to the PHAN-C pipeline which is available online, but our protocol using more recently published programs including epa-ng, gappa, and RaxML-ng.
Image Attribution
Image adapted from Fuchsman et al 2021 Environmental Microbiology 23: 2782-2800.
Guidelines
Keep a computer notebook that includes all your commands used and notes about results.
Use a text file, but do not use a Word file as Word messes up dashes and returns.
Materials
Install the following programs:
Biopython (Cock et al 2009)
epa-ng (Barbera et 2019)
gappa (Czech et al 2020)
Trimmomatic (Bolger et al 2014)
muscle (Edgar et al 2004)
Prokka (Seemann 2014)
MegaHIT (Li et al 2015)
RAxML version 8 (Stamatakis 2014)
RAxML-ng (Kozlov et al 2019)
PaPaRa 2.0 (Berger et al 2011, 2012)
All of these programs should be able to be installed with conda.
Before start
To utilize this pipeline, you will need to have a good computer that can use unix/linux terminal window. If you are using a laptop, you can greatly increase your storage capacity by having your metagenomic read blast databases on an external hard drive.
Naming and counting Metagenomic reads
Rename all the metagenome filenames so that they include
Depth_Cruise_Station
This can be done during the QC step, or by hand
QC with Trimmomatic (Bolger et al 2014)
Trimmomatic information and downloads can be found here: http://www.usadellab.org/cms/?page=trimmomatic
You will need an adaptor file TruSeq3-PE-2.fa that you can download with Trimmomatic
Name the output files with Depth_Cruise_Station
Command
trims adaptors and removes bad reads
Trimmomatic
The result of this command are 4 fastq files: 2 with paired end reads and 2 files where one paired read was deleted.
Convert fastq to fasta.
Note that we include both paried and unpaired reads in our read database, so also convert the unpaired read files here.
Information about the fastq_to_fasta script and how to download it can be found here:
This script does not work with .gz files; they must be unzipped.
The -n means to keep reads with Ns.
Command
converts fastq file to fasta
fastq_to_fasta
Note: keep your fastq files for assembly (step 7) and for other purposes. These fastq are also the files that you submit to NCBI SRA
Rename metagenomic reads in the fasta files. This script may need to be edited for your files. This script works on a whole folder of files. New metagenomic files will be created with _name.fasta at the end.
This script takes the Depth_Cruise_Station information from the metagenome filename and adds it to the name of each read. A dash (-) will be added between Depth_Cruise_Station and the original read name. This dash is used to separate and use the Depth_Cruise_Station information later. Use and underscore. Do NOT use normal dashes between Depth, Cruise and Station.
The one listed right below works with metagenomes downloaded from NCBI SRA.
Command
Add Depth, Cruise, and station to the metagenomic read names
The script can be found on GitHub
The one listed below works with metagenomes from the sequencing center.
Command
Add key information to each read
new command name
The script can be found on GitHub
Remember to include the pathway to the folder where you are storing the script in the command.
Use script Counting_whole_folder.py to count the number of reads in the file, for every metagenome in a folder. In this script, change the outfile name if desirable.
This script outputs a text file with the name of the metagenome and the number reads in the file.
Command
Counting metagenomic reads of a folder full of metagenomes
Remember to include the pathway to the folder where you are storing the script in the command.
The script can be found on GitHub
Use the read counts to create normalization factors.
First open the text file of read counts in excel. Add together forward and reverse read numbers.
In the Fuchsman lab, we use the ETNP 100 m sample to normalize all other samples in our collection: 485556135 reads
normalization factor for Z = 485556135 / reads in sample Z
The names need to be Depth_Cruise_Station without the .fasta or other extras
A: sample name, B: total reads, C: normalization factor
| A | B | C | |
| 100m_HOT_272 | 33647796 | 1.443070298 | |
| 125m_HOT_272 | 91158758 | 0.532654635 | |
| 150m_HOT_272 | 75113107 | 0.646440241 | |
| 175m_HOT_272 | 16552301 | 2.933497584 | |
| 200m_HOT_272 | 83220174 | 0.583465915 | |
| 225m_HOT_272 | 80424227 | 0.603750099 | |
| 250m_HOT_272 | 124980536 | 0.388509576 | |
| 25m_HOT_272 | 25193222 | 1.92734915 | |
| 45m_HOT_272 | 66381762 | 0.731468005 |
This file should be saved as a .csv, and then it should be saved with UNIX line breaks. (We do this in BBEDIT.)
This file is used in by the normalization script at the end of the phytogenetic read placement pipeline.
Assemble proteins from reads
Use fastq files to assemble contigs using MEGAHIT (Li et al 2015). We usually assemble each depth separately.
If you are looking for rare taxa, a combined assembly of similar samples can help.
Find and install MEGAHIT from here:
Command
Assemble contigs with megahit
new command name
Remove contigs less than 1000 basepairs (or whatever cut off you choose)
The script can be found on GitHub
Command
Cut short contigs
new command name
Remember to include the pathway to the folder where you are storing the script in the command.
Script credit: Ryan Groussman
Annotate contigs with prokka (Seemann, 2014). Prokka calls genes/proteins and annotates them.
Find prokka and information about prokka here:
Prokka has version dependencies. We keep prokka in its own conda environment.
Prokka only allows names that are <20 characters
This script changes names in a MegaHIT contig.fasta file to <20 characters
Change the infile and outfile names in the script
Command
Names for prokka
new command name
Remember to include the pathway to the folder where you are storing the script in the command.
To run prokka, first we activate the prokka conda environment
Command
Activate prokka environment
new command name
Then run the following command
Command
Run prokka
new command name
Prokka numbers the proteins. The following script links the proteins back to their original contigs. It adds the contig name to the beginning of the protein name. The table file from prokka is needed to run this script.
It also adds an identifier before the contig name that indicates the place the original metagenome was from.
Example: ETNP_1000m_contig#_prokka#
The script accession_dict_name_change.py is on GitHub
Command
Link protein to contig
new command name
Remember to include the pathway to the folder where you are storing the script in the command.
The exact same script can be used to link genes to their original contigs. Just use the gene file from prokka instead of the protein file.
Making local Blast Databases
Install Blast+ (Altschul et al.,1997)
I recommend installing all software with conda
See directions here: https://conmeehan.github.io/blast+tutorial.html
Make a nucleotide blast database for the metagenomic reads
Put all the metagenomes from one station into one blast database.
(if the blast database becomes too large, we end up having problems)
-hash_index and -parse_seqids are needed in the command for the parse script to work later.
Command
Making a metagenomic read BLAST database
new command name
Make a protein blast database for proteins from assembled metagenomes
We usually make one protein blast database per cruise.
-hash_index and -parse_seqids are needed in the command for the parse script to work later.
Use the renamed proteins from step 9.3.
Command
Assembled protein database
new command name
Making phylogenetic trees
Collect all available relevant reference sequences from NCBI (or public database of choice) that you want on your tree. Make sure your tree has outgroups which include other closely related genes or organisms.
Also choose some sequences across the diversity of the tree to be "seeds" for blasting and place these in a "seed" text file.
Blast your database of assembled proteins using the seed file
For this we use e values between -60 and -100, depending on the desired results. If we are looking at a particular organisms, we might use an e value of -100. If we are looking at the whole community, an e value of -60 allows broader results.
Command
Blast database of proteins for adding sequences to a phylogenetic tree.
Blast protein database
This output is not user friendly so we must use the following parse script to be able to get a useable output.
Here is a link to the Parse script used to make the output useable. This script uses python 2.
Script credit: Cedar McKay
For python 3, use:
Script credit: Cedar McKay and the PHAN-C pipeline
Both scripts use the same inputs. For more information about this script, read:
This script uses Biopython (Cock et al 2009)
Command
Parse the blast output file
Parse script command
In this command, c is the number of threads.
-e is the path for the blast database.
Keep track of output in the terminal window which should look like:
1310 of 2264 were unique hits (57.86%)
The script creates 3 files. This time we want the blast.best.hits_all.fasta, which is a list of non-redundant fasta files. .
The blast.best.hits_all.fasta file can then be added to the reference sequence fasta file.
Remove illegal characters
In order for sequences to be used in the making of the phylogenetic tree, a number of character types are not permitted including:
".", ",", ".", " ", "|", "[", "(", ";", ":"
Replacing with underscores is often the best way to not lose information in the sequence name.
Align sequences using muscle (Edgar, 2004)
Command
Muscle command
Visualize your alignment with Seaview (or program of your choice)
Are there any wonky sequences? 1) Sequences that do not align with the other sequences, 2) extremely short sequences, 3) sequences with long "introns" or unaligned segments. You may want to remove these sequences, or see if they are reverse and complement.
Then run muscle again on the corrected file.
Make a non-redundant alignment file or a "reduced file"
RaxML version 8 (Stamatakas et al 2014) can be downloaded here:
Use RaxML to pretend to make a phylogenetic tree. Then stop the command after a reduced alignment file is created.
Command
Obtain Reduced file
The format of the reduced file is necessary to get PaPaRa to work later.
Make a preliminary phylogenetic tree using RaxML-ng (Kozlov et al 2019)
Download RaxML-ng here:
https://github.com/amkozlov/raxml-ng (or use conda)
Different command are used for an amino acid tree versus a nucleotide tree
For an nucleotide tree:
Command
Make Preliminary Nucleotide Phylogenetic tree
For amino acid tree:
Command
Make Preliminary Amino Acid Tree
Look at your tree quality. Do you have some extremely long branch lengths? If so, you may want to remove those sequences from the tree, or find more sequences that are similar to the strange sequence. Usually we remove. Start over with muscle (Step 17).
If the assembled proteins form clusters without references, I recommend blasting representative proteins against NCBI to find closest hits. Add these hits to your reference file, make sure they do not have illegal characters, and start over with muscle (Step 17).
Bootstrap the tree with tbe bootstraps
When you are happy with your tree, use RaxML-ng to bootstrap it using Transfer Bootstrap Expectation
Read about Transfer Bootstrap Expectation here (Zaharias et al 2023):
Command
Bootstrap phylogenetic tree
If you are bootstrapping a nucleotide tree, the data type is DNA.
Bootstrapping can take awhile and can take a lot of cores. This is a step where you might consider using HPC server.
Metagenomic Read Placement
Use your seed file to blast your metagenomic read database created in step 11.
This seed file is usually composed of amino acid sequences to allow the widest range of hits.
However, for genes like 16S rRNA, ITS or 18 rRNA the seed file must be nucleotide.
Here we use an evalue of -5 to get the broadest range of reads possible.
Command
Blast metagenomic read database with amino acid seed file
Command
Blast metagenomic read database with nucleotide seed file
Parse this blast file as in step 15.
This time we want the best.hits file because it contains information on the direction of the read-- whether or not it needs to be reverse and complemented
Trim and Reverse and Complement
This step trims reads that partially contain a different gene, reverse and compelements if necessary, removes all reads with Ns, and translates to amino acids
Jaci Saunders wrote this script.
trim_reads_format_for_alignment_specify_minLength.py can be found:
A different version of the same script can be found in the PHAN-C pipeline
Command
Trim and format reads
Trim and format Metagenomic Reads
-m 100 means that all reads shorter than 100 basepairs are deleted
There are many output files from the script. If you have an amino acid tree, you want the "AA_translation_stops_removed_BEST_for_PAPARA.fasta" output file.
If you have a nucleotide tree, you want the "names_formatted_query_seqs_NT.fasta" file
Align metagenomic reads to the reference sequences used to make the phylogenetic tree using PaPaRa (Berger and Stamatakis, 2011)
PaPaRa 2.0 can be found here:
Command
PaPaRa for amino acids
Command
PaPaRa for nucleotides
Note that the -a indicates amino acids
-j is the number of threads
Combine paired end reads.
A long gene will have more forward and reverse paired reads than a short gene. To be able to compare between genes properly, we merge the alignments of paired end reads and count them as one.
If you have downloaded metagenomes from SRA, use spacejoin_Geotraces.py found here
If you are using your own metagenomes, use spacejoin.py found here:
spacejoin.py was written by Jaci Saunders
both use the same command.
Command
Link paired end reads
Spacejoin
-r is the number of references sequences composing the tree.
Adjust formatting. This step is not always necessary, but we always do it to prevent errors later on in the pipeline.
phyml2fasta_space_dash.py can be found here:
Adjust the input and output file names inside the script.
Split aligned file into references and reads to use in epa-ng
Use the spacedash file that you just formated
NR < (the number of references from spacejoin * 2)+1
So if you have 492 references in your tree, the number to use here is 985
Command
Split between references and reads
Place metagenomic reads on the tree using epa-ng (Barbera et al 2019)
epa-ng can be found here:
-s is the reference only file
-t is the tree file
-q is the reads only file
Command
EPA-ng command for pipeline
EPA-ng
the filter-max 1 is important so that we don't count the same read multiple times.
It means, only place the read once on its best spot.
If you are using a nucleotide tree, the model is: GTR
If you are using an amino acid tree, the model is: WAG (or whatever other model you used to make the tree)
Cut reads with long pendant lengths (>2) to prevent false assignments
Ideally this script will only cut a very small number of reads. If this step cuts many reads, it is a warning sign that your phylogenetic tree is missing key sequences/outgroups.
The pendant length used to cut is adjustable, but we recommend 2
the script jplace_file_cutter.py can be found here:
Adjust the name of the outfile in the script.
The script will print:
"The number of reads above length 2 is...#" -- keep track of this number in your notebook
Make a taxon file to use with gappa
Making the taxon file is one of the most important steps of the pipeline. The Information that you obtain at the end of the pipeline is directly related to what you designate here.
open your tree in fig tree and export to as a pdf.
In Illustrator or equivalent, mark the taxonomic groups of interest on the phylogenetic tree
--determine the taxonomic splits e.g. ecotypes, outgroups, phages vs hosts
Make a new text file with the name of the reference on the tree and the classification seperated by a tab
The names must match the names on the tree exactly.
You can copy the reference name, or a whole section of names, in FigTree and paste it into the text file
The taxon section can have multiple levels separated by a semicolon.
Example:
pelagiphage_ref_AP014234 (tab) Virus;Pelagiphage;Podopelagiphage group1
In this example, one read will show up in our final counts under three headings: virus, pelagiphage, and podopelagiphage group 1
You must spell these groups exactly the same way for all reference sequences in the same group
Every reference sequence in the tree must be in the taxon file
If you mess up and either have two tabs on the same line or zero tabs, gappa will not work.
If gappa gives you a tab error:
tab.py is a script to find the line where you either have no tab or two tabs
find the script here:
Inside the script, designate your taxon file
Assign Taxonomy with gappa (Czech et al 2020)
gappa can be found here:
Command
Run gappa with taxon file
Run gappa
The out-dir is the name of a folder that will be created
Count reads for each taxonomy for each metagenomic sample separately
From inside the gappa output folder, use seperate_by_sample_and_phylotype_comma_CLARA.py found at:
Adjust the outfile name in the script
Normalization by gene length and sample sequencing depth
The script normalize_for_post_processing_zeros_added3.py can be found at:
To use this script, you will have to change in the body of the script
-- the outfile name
-- a file of normalization factors (see step 6 and use that format)
-- the gene length in nucleotide basepairs
-- the "for post processing" file created in step 32. This file is in the script twice.
The file produced by this script is our final product that can be used in excel or R.
Extras
To pull out metagenomic reads that placed within a specific taxonomy on the phylogenetic tree
Reasons to do this: If you originally place reads on a very large broad tree, but then want to replace a subset of the reads on a more detailed subtree.
The scrpt Pulling_out_Stramenopile.py can be found at:
Inside the script, make the following changes:
Use the exact name used in the taxon file for the group of interest for Taxonomy
The fasta_file is the file of reads created in step 27 and used with epa-ng in step 28
The per_query file was created by gappa in step 31 and used in step 32.
To replace these reads on a new tree, start with PaPaRa at step 24.
Finding the % similarity of reference sequences in a taxon groups on the phylogenetic tree
The script percent_similarity_script.py can be found at:
This script uses Biopython.
To run this script, you need to make a list of the taxon groups of interest in a text file. These taxa are the ones where % similarity will be calculated
You will need the taxon file created in step 30.
You will need the text file of reference sequences used to make the tree (fasta_file)
Protocol references
1. Altschul, S. F., Madden, T. L., Schäffer, A. A.,
Zhang, J., Zhang, Z., Miller, W., & Lipman, D. J. (1997). Gapped BLAST and
PSI-BLAST: a new generation of protein database search programs. Nucleic
Acids Res., 25, 3389–3402.
2. Barbera, P., Kozlov, A. M., Czech, L., Morel, B., Darriba, D., Flouri, T.,
& Stamatakis, A. (2019). EPA-ng: Massively Parallel Evolutionary Placement
of Genetic Sequences. Systematic Biology, 68(2), 365–369.
https://doi.org/10.1093/sysbio/syy054
3. Berger, S. A., & Stamatakis, A. (2011). Aligning short reads to
reference alignments and trees. Bioinformatics, 27(15),
2068–2075. https://doi.org/10.1093/bioinformatics/btr320
4. Berger, S. A., & Stamatakis, A. (2012). PaPaRa 2.0: a vectorized
algorithm for probabilistic phylogeny-aware alignment extension.Institute
for Theoretical Studies, Retrieved from http://sco.h-its.org/exelixis/pubs/Exelixis-RRDR-2012-5.pdf
5. Bolger, A. M., Lohse, M., & Usadel, B. (2014). Trimmomatic: a flexible
trimmer for Illumina sequence data. Bioinformatics, 30(15), 2114–2120.
https://doi.org/10.1093/bioinformatics/btu170
6. Czech, L., Barbera, P., & Stamatakis, A. (2020). Genesis and Gappa:
processing, analyzing and visualizing phylogenetic (placement) data. Bioinformatics,
36(February), 3263–3265. https://doi.org/10.1093/bioinformatics/btaa070
7. Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high
accuracy and high throughput. Nucleic Acids Res, 32, 1792–1797.
8. Fuchsman, C. A., Garcia Prieto, D., Hays, M. D., & Cram, J. A. (2023).
Associations between picocyanobacterial ecotypes and cyanophage host genes
across ocean basins and depth. PeerJ, 11, e14924.
9. Kozlov, A. M., Darriba, D., Flouri, T., Morel, B., & Stramatakis, A.
(2019). RAxML-ng: a fast, scalable and user-friendly tool for maximum
likelihood phylogenetic inference. Bioinformatics, 35, 4453–4455.
10. Li, D., Liu, C. M., Luo, R., Sadakane, K., & Lam, T. W. (2015).
MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics
assembly via succinct de Bruijn graph. Bioinformatics, 31(10),
1674–1676. https://doi.org/10.1093/bioinformatics/btv033
11. Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics,
30(14), 2068–2069. https://doi.org/10.1093/bioinformatics/btu153
12. Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis
and post-analysis of large phylogenies. Bioinformatics, 30,
1312–1313. https://doi.org/10.1093/bioinformatics/btu033
13. Zaharias, P., Lemoine, F., & Gascuel, O. (2023). Robustness of
Felsenstein’s Versus Transfer Bootstrap Supports With Respect to Taxon
Sampling. Systematic Biology, 72(6), 1280–1295.
https://doi.org/10.1093/sysbio/syad0521.