Apr 23, 2024
Oxford Nanopore Long-Read Sequencing for Identification of Somatic Variants in Tumor/Normal Pairs
This protocol is a draft, published without a DOI.
- Isabella Livingston1
- 1North Carolina State University



Protocol Citation: Isabella Livingston 2024. Oxford Nanopore Long-Read Sequencing for Identification of Somatic Variants in Tumor/Normal Pairs. protocols.io https://dx.doi.org/
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: April 19, 2024
Last Modified: April 23, 2024
Protocol Integer ID: 98418
Keywords: metagenomics, Nanopore sequencing, WGS
, Cancer Genetics, somatic variants in tumor sample, somatic variants in tumor, read sequencing for identification, oxford nanopore long, normal pairs oxford nanopore technology, tumor sample, read sequencing, sequencing data, somatic variant, sequencing, streamlined workflow for the identification, epi2me, analysis with the epi2me, tumor
Disclaimer
This was created as part of a class assignment. It is based on published and well-documented protocols (referenced)
Abstract
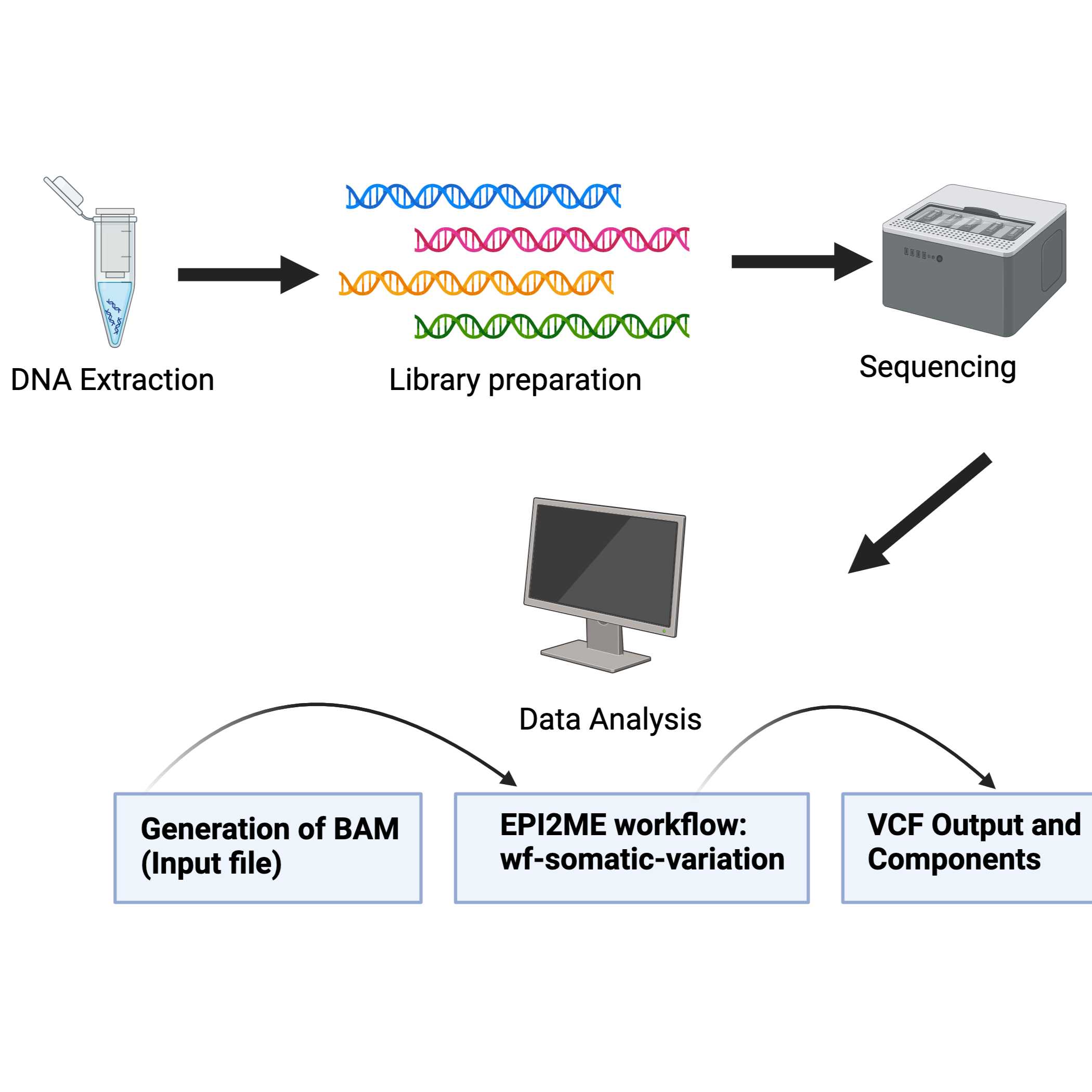
Oxford Nanopore Technologies (ONT) offers a streamlined workflow for the identification of somatic variants in tumor samples. This workflow generates long-read and high-quality sequencing data that can be used for analysis with the EPI2ME wf-somatic-variation workflow. This workflow can be run through a user interface or the command line and minimizes hands-on analysis.
Image Attribution
Figure created with BioRender.com
Guidelines
All Centrifuge steps are done at room temperature (15-25°C)
Vortexing should be done by pulse-vortexing for 5 - 10 Seconds
Check kit specifications for other potentially hazardous material
Optional: Samples can be treated with RNAse A to digest RNA during the procedure (not supplied in listed DNA extraction kit)
To multiplex libraries, combine with the ONT Native Barcoding Kit
The recommended workflow for identifying somatic variants in tumor-normal sequencing data is the wf-somatic-variation through EPI2ME. This can be run through the GUI (recommended) or command line.
For other modified options including indels and methylation analysis refer to : https://github.com/epi2me-labs/wf-somatic-variation
Materials
- 1 ug (or 100-200 fmol) high molecular weight genomic DNA
- Or 100+ ng high molecular weight genomic DNA if performing DNA fragmentation
- Ligation Sequencing Kit V14 (SQK-LSK114)
Consumables Required:
- MinION and GridION Flow Cell
- Qubit dsDNA HS Assay Kit (Invitrogen, Q32851)
- NEBNext Companion Module for ONT Ligation Sequencing (NEB, E7180S or E7180L)
or
- NEBNext FFPE Repair Mix (NEB, M6630)
- NEBNext Ultra II End repair/dA-tailing Module (NEB, E7546)
- NEBNext Quick Ligation Module (NEB, E6056) (Optional)
- Bovine Serum Albumin (BSA) (50 mg/ml) (e.g Invitrogen‱ UltraPure‱ BSA 50 mg/ml, AM2616)
- Freshly prepared 80% ethanol in nuclease-free water
- Nuclease-free water (e.g. ThermoFisher, AM9937)
- 1.5 ml Eppendorf DNA LoBind tubes
- 0.2 ml thin-walled PCR tubes
- Qubit‱ Assay Tubes (Invitrogen, Q32856)
Equipment Required
- MinION or GridION device
- MinION Flow Cell Light Shield
- Hula or other gentle rotator mixer
- Magnetic rack
- Microfuge
- Vortex Mixer
- Thermal Cycler
- P1000, P200, P100, P20, P10, P2 pipettes and tips
- Ice bucket
- Timer
- Fluorometer (Qubit or other for QC check)
Safety warnings
Buffers may contain hazardous chemicals. Check SDS before use.
Ethics statement
Not ethical concerns to disclose
Before start
Buffer ATL and Buffer AL may form precipitates upon storage. If necessary, warm to 56°C until the precipitates have fully dissolved. Buffer AW1 and Buffer AW2 are supplied as concentrates. Before using for the first time, add the appropriate amount of ethanol (96–100%) as indicated on the bottle to obtain a working solution. Preheat a thermomixer, shaking water bath or rocking platform to 56°C for use in step 2.
DNA Extraction (QIAGEN DNEasy Blood and Tissue Kit or alternative extraction method)
Add 180 ul Buffer ATL into a labeled 1.5 mL tube for each tissue specimen. Cut 25 mg of tissue into small pieces and place in the labeled tube.
Spin down the samples and add 20 ul Proteinase K. Mix by vortexing and incubate at 56°C until tissue is completely lysed. Vortex occasionally to check status.
(Optional): Add 4 ul RNase A (100 mg/ml), mix by vortexing, and incubate for 2 min at room temperature.
Vortex for 15 seconds. Add 200 ul Buffer AL to the sample and mix thoroughly by vortexing. Add 200 ul ethanol (96%-100%), and mix again by vortexing.
It is essential that the sample, Buffer AL, and ethanol are mixed immediately and thoroughly by vortexing or pipetting to yield a homogeneous solution. Buffer AL and ethanol can be premixed and added together in one step to save time when processing multiple samples. A white precipitate may form on addition of Buffer AL and ethanol. This precipitate does not interfere with the DNeasy procedure. Some tissue types (e.g., spleen, lung) may form a gelatinous lysate after addition of Buffer AL and ethanol. In this case, vigorously shaking or vortexing the preparation is recommended.
Pipet the mixture into the provided DNeasy Mini spin column in a 2 ml collection tube. Centrifuge at top speed for 1 minute. Discard the flow-through and collection tube.
Place the spin column in a new 2 ml collection tube, add 500 ul Buffer AW1 and centrifuge for 1 min at top speed. Discard flow-through and collection tube.
- Place the spin column in a new 2 ml collection tube, add 500 ul Buffer AW2, and centrifuge for 1 minute at top speed. Discard flow-through but retain collection tube.
Add 500 ul of 70% (cold) ethanol. Centrifuge for 3 minutes at top speed to dry the DNeasy membrane. Discard flow through and collection tube
Remove the spin column carefully so it does not come into contact with the flow-through.
This will result in the carry-over of ethanol.
Place the spin column in a new 2ml collection tube and add 200 ul Buffer AE directly onto the membrane. Let sit at room temperature for 1 minute and then centrifuge for 1 min at max speed to elute. Repeat the elution step to increase DNA yield.
Proceed to quantify the DNA.
Library Preparation (ONT Ligation Sequencing Kit): Note third-party consumables are required
- Thaw the DNA Control Sample (DCS) at room temperature, spin down, mix by pipetting, and store on ice until ready to use
Recommended but 1 ul sample DNA or nuclease-free water can be used in place
Prepare the NEBNext FFPE DNA Repair Mix, and NEBNext Ultra II End Repair/ dA-tailing Module reagents according to manufacturer’s recommendations. Store on Ice
a. Recommended to flix or invert the reagent tubes to mix
b. Do not vortex the FFPE DNA Repair Mix or Ultra II end Prep Enzyme Mix
Prepare the DNA in nuclease-free water:
1) Transfer 1 ug DNA into a 1.5 ml Eppendorf DNA LoBind tube
2) Adjust the volume to 47 ul with nuclease-free water
3) Mix by pipetting up and down gently or by flicking the tube
4) Spin down briefly in a microfuge
In a 0.2 ml thin-walled PCR tube, mix the following: (A = Reagent)(B = Volume ul)
| DNA from Step 3 | 47 | |
| DNA CS (optional) or nuclease-free water | 1 | |
| NEBNext FFPE DNA Repair Buffer | 3.5 | |
| NEBNext FFPE DNA Repair Mix | 2 | |
| Ultra II End-prep Reaction Buffer | 3.5 | |
| Ultra II End-prep Enzyme Mix | 3 | |
| Total | 60 ul |
Between additions of each reagent, pipette to mix 10-20 times (gently) ReagentVolume (ul)DNA from Step 347DNA CS (optional) or nuclease-free water1NEBNext FFPE DNA Repair Buffer3.5NEBNext FFPE DNA Repair Mix2Ultra II End-prep Reaction Buffer3.5Ultra II End-prep Enzyme Mix 3Total60 ul
Mix thoroughly by gently pipetting and spinning down
Using a thermal cycler incubate with the following conditions
20°C for 5 minutes
65°C for 5 minutes
Resuspend Ampure XP beads by vortexing
Transfer the DNA sample to a new clean 1.5 ml Eppendorf DNA LoBind tube
Add 60 ul of resuspended AmPure Beads to the end-prep reaction. Mix by flicking or inverting the tube
Incubate on hula or other rotator for 5 minutes at room temperature
Prepare 500 ul of fresh 80% ethanol in nuclease-free water
Spin down the sample and pellet on a magnet until the supernatant is clear and colorless. Keep the tube on the magnet, and pipette off the supernatant
Keep the tube on the magnet and wash the beads with 200 ul of freshly prepared 80% ethanol without disturbing the pellet. Let sit for 30 seconds. Remove the ethanol using a pipette and discard.
Repeat the wash
Spin down and place the tube back on the magnet. Pipette off any residual ethanol. Allow to dry for ~30 seconds. Do not dry the pellet to the point of cracking
Remove the tube from the magnetic rack and resuspend the pellet in 61 ul nuclease-free water. Incubate for 2 minutes at room temperature
Pellet the beads on a magnet until the eluate is clear and colorless (~ 1 min or more)
Remove and retain 61 ul of eluate into a clean 1.5 ml Eppendorf DNA LoBind tube
Use 1 ul of eluted sample using a fluoremeter
Spin down the Ligation Adapter (LA) and Quick T4 Ligase. Store on ice
Thaw Ligation Buffer (LNB) at room temperature, spin down and mix by pipetting. Store on ice immediately after thawing and mixing
Due to viscosity, vortexing this buffer is ineffective
Thaw the Elution Buffer (EB) at room temperature. Mix by vortexing. Spin down and store on ice
Thaw either the Long Fragment Buffer (LFB) or Short Fragment Buffer (SFB) at room temperature and mix by vortexing. Then spin down and place on ice
In a 1.5 ml Eppendorf DNA LoBind tube, mix in the following order:
Between addition of each reagent, pipette to mix (gently) 10 - 20 times
| Reagent | Volume (ul) | |
| DNA Sample from the previous step | 60 | |
| Ligation Buffer (LNB) | 25 | |
| NEBNext Quick T4 DNA Ligase | 10 | |
| Ligation Adapter (LA) | 5 | |
| Total | 100 ul |
Table adapted from the SQK-LSK114 Protocol
Mix thoroughly by gently pipetting and briefly spinning down.
Incubate the reaction for 10 minutes at room temperature
Resuspend the AMPure XP Beads (AXP) by vortexing
Add 40 ul of resuspended AMPure XP Beads (AXP) to the reaction and mix by flicking the tube
Incubate on a Hula mixer for 5 minutes at room temperature
Spin down the sample and pellet on a magnet. Keep the tube on the magnet, and pipette off the supernatant when clear and colorless.
Wash the beads by adding either 250 ul Long Fragment Buffer or 250 ul Short Fragment Buffer. Flick the beads to resuspend, spin down, return the tube to the magnet and allow the beads to pellet. Remove the supernatant and discard
Repeat
Spin down and place the tube back on the magnet. Pipette off any residual supernatant. Allow to dry for ~30 seconds
Do not dry the pellet to the point of cracking
Remove the tube from the magnet and resuspend the pellet in 15 ul Elution Buffer (EB). Spin down and incubate for 10 minutes at room temperature.
For high molecular weight DNA, incubating at 37°C can improve the recovery of long fragments
Pellet the beads on a magnet until the emulate is clear and colorless (~1 min or more)
Remove and retain 15 ul of eluate containing the DNa library into a clean 1.5 ml Eppendorf DNA LoBind tube. Dispose of the pelleted beads
Proceed to quantify 1 ul of eluted sample using a fluorometer
Depending on required output, prepare the final library to 35-50 fmol for high output of simplex data, or 10-20 fmol for duplex data, in 12 ul of Elution Buffer (EB).
If library yields are below input recommendations, load the entire library.
Recommended to use duplex data
Load 10 - 20 fmol of final library. Loading more than 20 fmol of DNA can reduce the rate of duplex read capture
Proceed to priming and loading of the flow cell
Data Analysis using EPI2ME
Following the sequencing run, BAM files (the required input for the workflow) can be generated from the POD5 or FAST5 files following the wf-basecalling workflow
Alternatively, BAM files can be generated from fastq files using the wf-alignment workflow
Both workflows generate aligned BAM files that are compatible with the wf-somatic-variation workflow.
If no matched normal sample is available, the tumor-only mode must be used.
QC of sequencing data and pre-processing
Computes depth of sequencing of the BAM files (mosdepth)
Computes read alignment statistics for each BAM (fastcat)
Other commands
--tumor_min_coverage :used to determine whether the input tumor BAMs have a depth greater than this value
--normal_min_coverage :used to determine whether the input normal BAMs have a depth greater than this value
Both steps can be skipped if the minimum coverage level is set to zero
Somatic Variant Calling (short variants) using ClairS
Version implemented by workflow is ClairS (v0.1.6) which is used to identify somatic variants in paired tumor-normal samples
Supported basecalling models can be found on https://github.com/epi2me-labs/wf-somatic-variation)
Clair3 is used in this workflow to call germline variants in both the tumor and normal sample
These are used to refine the somatic variant calling
Options for reducing computational demands
- - fast_mode: reduces accuracy of variant calling
- -normal_vcf: provide a pre-computed VCF file with germline calls for the normal sample
- - germline false: disables the germline calling process
Calling Somatic structural variants (SVs) with Nanomonsv
Workflow calls somatic structural variants with long-read sequencing data. It starts from paired tumor/normal samples and will:
Parse signatures of structural variants using nanomonsv parse
Call the somatic SVs with nanomonsv get
(Optional) Filters the SVs out in simple repeats with add_simple_repeat.py
(Optional) Annotate transposable and repetitive elements with nanomonsv insert_classify
Protocol references
- Qiagen INC. DNeasy Blood & Tissue Kit. https://www.qiagen.com/us/Resources/ResourceDetail?id=d155700e-edce-4fb1-9f97-8faa3265ec8e&lang=en
- https://community.nanoporetech.com/docs/prepare/library_prep_protocols/genomic-dna-by-ligation-sqk-lsk114/v/gde_9161_v114_revu_29jun2022
- https://nanoporetech.com/sites/default/files/s3/literature/tumour-normal-workflow.pdf
- Github. Wf-somatic-variation. https://github.com/epi2me-labs/wf-somatic-variation
- Luo, R., Wong, CL., Wong, YS. et al. Exploring the limit of using a deep neural network on pileup data for germline variant calling. Nat Mach Intell 2, 220–227 (2020). https://doi.org/10.1038/s42256-020-0167-4
- Yuichi Shiraishi, Junji Koya, Kenichi Chiba, Ai Okada, Yasuhito Arai, Yuki Saito, Tatsuhiro Shibata, Keisuke Kataoka, Precise characterization of somatic complex structural variations from tumor/control paired long-read sequencing data with nanomonsv, Nucleic Acids Research, Volume 51, Issue 14, 11 August 2023, Page e74, https://doi.org/10.1093/nar/gkad526