Feb 17, 2026
Ni-NTA spin-down method for purifying large proteins (batch method)
This protocol is a draft, published without a DOI.
- Ainsley Lederer1
- 1University of Pittsburgh



Protocol Citation: Ainsley Lederer 2026. Ni-NTA spin-down method for purifying large proteins (batch method). protocols.io https://dx.doi.org/
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: December 05, 2025
Last Modified: February 17, 2026
Protocol Integer ID: 234328
Keywords: purifying large protein, batch method purification, large scale protein isolation, protein in the elution step, large protein, protein, batch method
Abstract
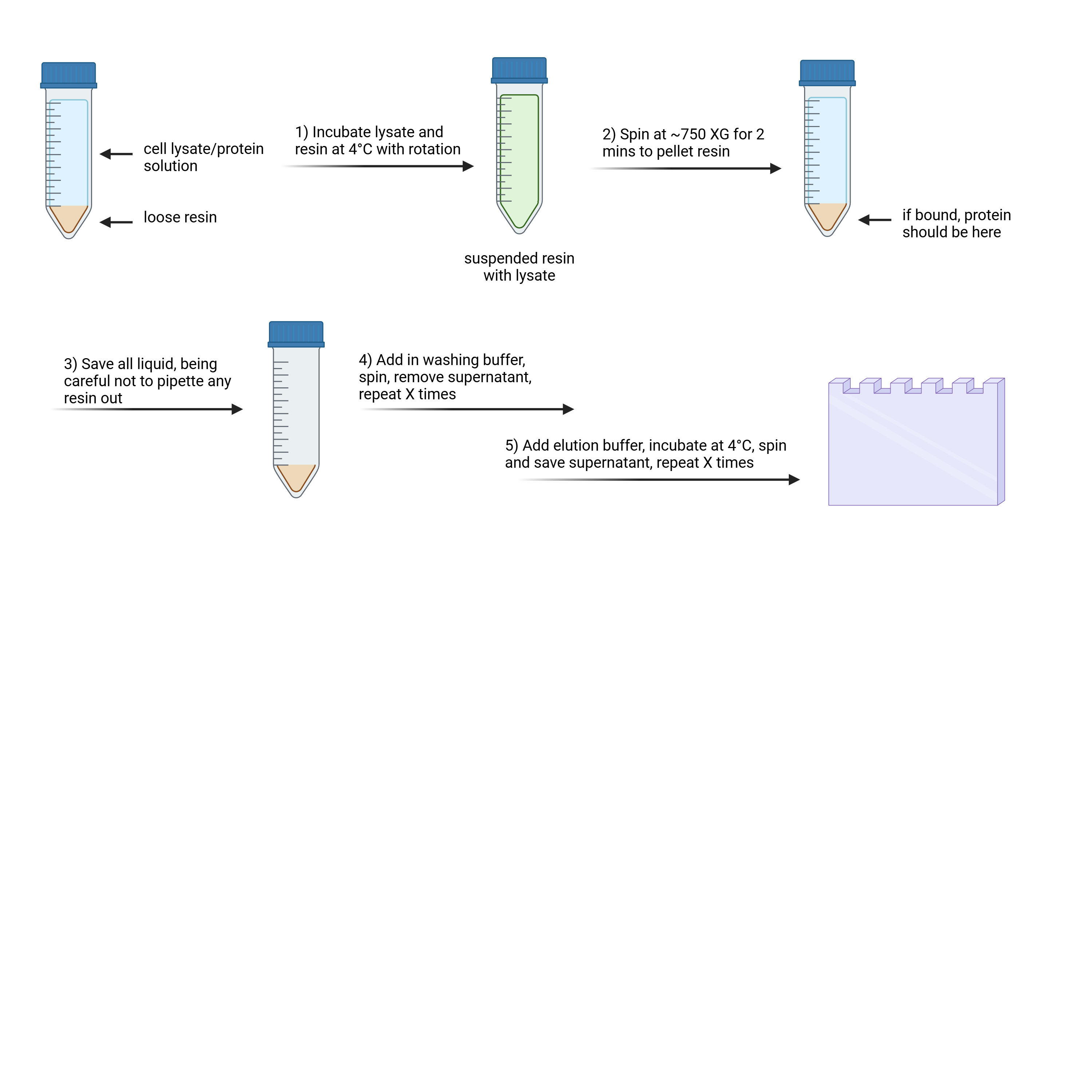
This protocol details how to perform batch method purification, using "loose" resin (i.e., not a packed column). The protocol is written for large scale protein isolation, for example by using lysate from 4 L E. coli culture. That said, it can be easily scaled depending on your needs!
The expected result from this protocol would be a gel showing the input sample, supernatant ("flow through") from incubation, binding step, washing step, and elution step. If your protein has bound to the resin, you should see the protein in the elution step of this process.
Guidelines
The framework of this protocol can be applied to other resins and sample volumes too! I have had success using this method on both the small-scale (microcentrifuge tube) and the large scale, as detailed here. Other resins I have used successfully include Protein A resin, glutathione resin, and Streptactin XT resin.
Always look at the manual for your specific resin before performing this protocol, and change the buffers & incubation times as needed!
Note that many manuals may say their resin is not suited for batch method purification. In my experience, this is typically because they want you to feel like you have to purchase their special centrifugal columns. Regardless, if you are concerned, you should do a small-scale test first in a microcentrifuge tube using this protocol.
Materials
Ni-binding buffer: 50 mM Tris-HCl pH 7.5, 100 mM NaCl, 10 mM imidazole, 10% glycerol, 5 mM beta-mercaptoethanol (BME)
Ni-washing buffer: 50 mM Tris-HCl pH 7.5, 100 mM NaCl, 20 mM imidazole, 10% glycerol, 5 mM BME
Ni-elution buffer: 50 mM Tris-HCl pH 7.5, 100 mM NaCl, 250 mM imidazole, 10% glycerol, 5 mM BME
Safety warnings
*DO NOT DISCARD USED RESIN – SAVE IN A TUBE FOR WASHING LATER*
Before start
Batch method purification can be frustrating at first, because it is easy to disturb and resuspend the resin when removing your sample from the tube. But always remember than you can spin the tube down again if this happens! Patience is key. When starting out, save all your supernatants, so you can recover lost resin later!
Resin washing & equilibration
Pipette the appropriate amount of resin in 50 mL falcon tubes. Different resin brands have different capacities, but generally we use 2 mL resin slurry per ~15 mL E. coli cell lysate. It is helpful to cut the end of a 1 mL tip for pipetting resin.
Spin down the resin slurry at 750xG for 2 minutes in swing bucket. Carefully pipette off supernatant.
Add ~5 RV (packed resin volume) of Ni-binding buffer, invert to mix until all resin is resuspended, then spin at 750xG for 2 mins and discard supernatant. A swing bucket rotor is preferable to a fixed-angle rotor. Supernatant should be discarded by using a serological pipette first to remove majority of the liquid, then a micropipette to remove the rest, without disturbing the resin. This takes some practice to do properly! If the resin is disturbed, just spin the tube down again!
If you are new to this technique, I recommend saving all supernatant, even from these washing steps. This is for 2 reasons: 1) the resin will settle to the bottom of the tube and you can assess your technique, and 2) you can recover this lost resin later using a gravity column or similar.
Note
A note on some of the abbreviations I use: 2 mL resin slurry = 1 mL packed resin (because the slurry is 50% liquid, 50% resin). Check the resin bottle to see the suspension - for example, some resins are 20% resin, meaning you would need to use 5 mL of slurry to achieve 1 mL packed resin. For all steps, the amount of buffer used is dependent on the packed resin volume (RV). If you have 5 mL packed resin volume and the step says add 5 RV of Ni-binding buffer, then that means add 25 mL of buffer.
Repeat step 3 once more.
Lysate application and protein isolation
Add your cell lysate into the tube and invert to resuspend the resin. Reserve a few uL of lysate to run on a gel later. Incubate on the tube rotator for 30-60 mins at 4°C.
Spin the tubes at 750xG for 2 mins. Remove the supernatant using the method described in step 3. Save supernatant as "flow through" fraction.
Add ~8 RV of binding buffer, invert to mix several times. Spin at 750xG for 2 mins, save supernatant as the "binding" fraction.
Add ~8 RV of washing buffer, invert to mix several times. Spin at 750xG for 2 mins, save supernatant as "washing" fraction.
Add ~5 RV of elution buffer, and place on tube rotator in 4°C room. For Ni-NTA resin, I typically incubate the resin with elution buffer for ~15-30 minutes. Incubation time will vary depending on protein's affinity to resin.
Spin tube down at 750xG for 2 minutes, and carefully pipette as much supernatant as possible without also removing resin. I recommend removing as much liquid as possible, then spin the tube again, and see if you can get any more supernatant.
*DO NOT DISCARD USED RESIN – SAVE IN A TUBE FOR WASHING LATER*
Run your samples on a SDS-PAGE gel. I recommend to run the following samples:
1. Lysate (before application to beads)
2. "flow through"/supernatant from step 6
3. binding fraction, step 7
4. washing fraction, step 8
5. elution fraction, step 9
6. *10 uL resin, boiled in sample buffer for 5 minutes
*Use a P20 pipette and cut the tip, then pipette ~10 uL of resin after you've collected all of your elution fraction, and add ~15 uL sample buffer. This will allow you to see whether your protein has fully eluted, or if it is still stuck to the resin and requires another elution step.
Expected result
Gel showing results from batch-method purification of a large protein. Three washing steps were including, with one elution step. No sample of beads was loaded in this scenario after previously verifying this protein elutes quickly from the resin.