Dec 28, 2023
Native Barcoding (SQK-NBD114) gDNA for Adaptive Sampling using Oxford Nanopore Technologies
- 1Department of Genetics and Genomic Medicine, Great Ormond Street Institute of Child Health, University College London, London WC1N 1EH, UK.;
- 2Department of Genetics and Genomic Medicine, Great Ormond Street Institute of Child Health, University College London, London WC1N 1EH, UK;
- 3UCL Queen Square Institute of Neurology, 3rd Floor Queen Square House, London WC1N 3BG;
- 4Department of Neurodegenerative Disease, Queen Square Institute of Neurology, University College London;
- 5Department of Genetics and Genomic Medicine, Great Ormond Street Institute of Child Health, University College London;
- 6NIHR Great Ormond Street Hospital Biomedical Research Centre, University College London



Protocol Citation: claire.anderson , Hannah Macpherson, Emil Gustavsson, Jasmaine Lee, zhongbo.chen 2023. Native Barcoding (SQK-NBD114) gDNA for Adaptive Sampling using Oxford Nanopore Technologies. protocols.io https://dx.doi.org/10.17504/protocols.io.kxygx3qx4g8j/v1
Manuscript citation:
Chen, Z., Gustavsson, E.K., Macpherson, H., Anderson, C., Clarkson, C., Rocca, C., Self, E., Alvarez Jerez, P., Scardamaglia, A., Pellerin, D., Montgomery, K., Lee, J., Gagliardi, D., Luo, H., , Hardy, J., Polke, J., Singleton, A.B., Blauwendraat, C., Mathews, K.D., Tucci, A., Fu, Y.-H., Houlden, H., Ryten, M. and Ptáček, L.J. (2024), Adaptive Long-Read Sequencing Reveals GGC Repeat Expansion in ZFHX3 Associated with Spinocerebellar Ataxia Type 4. Mov Disord. https://doi.org/10.1002/mds.29704
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: December 04, 2023
Last Modified: May 31, 2024
Protocol Integer ID: 92032
Keywords: Native Barcoding, SQK-NDB114, Oxford Nanopore Technologies, ONT, Adaptive sampling, gDNA, genomic DNA, Targeted Long Read Sequencing, ASAPCRN, gdna for adaptive sampling, native barcoding of genomic dna, oxford nanopore technologies this protocol, using oxford nanopore technology, oxford nanopore technology, nanopore, gdna, time selection of dna molecule, rejection of dna molecule, dna molecule, genomic dna, native barcoding kit, dna, native barcoding, native barcoding kit 24 v14, adaptive sampling, sequencing
Funders Acknowledgements:
Leonard Wolfson clinical research fellowship to ZC
Grant ID: 157793
Fellowship Award from the Canadian Institutes of Health Research to DP
This work was supported in part by the Intramural Research Program of the National Institute of Health including: the Center for Alzheimer’s and Related Dementias, within the Intramural Research Program of the National Institute on Aging and the National Institute of Neurological Disorders and Stroke.
Grant ID: AG-3172
Medical Research Council
Grant ID: MR/S01165X/1, MR/S005021/1, G0601943, MR/V012177/1
Aligning Science Across Parkinson’s (ASAP) through the Michael J. Fox Foundation for Parkinson’s Research (MJFF)
Grant ID: ASAP-000478, ASAP-000509, ASAP-000520
Abstract
This protocol describes how to carry out native barcoding of genomic DNA (gDNA) using the Native Barcoding Kit 24 V14 (SQK-NBD114.24) for Adaptive Sampling using Oxford Nanopore Technologies (ONT). Adaptive Sampling is a method of real-time selection of DNA molecules for sequencing and rejection of DNA molecules that are not of interest by reversing the voltage across the pores.
Attachments



Guidelines
Native Barcoding Kit 24 V14 (SQK-NBD114.24) should only be used in combination with R10.4.1 flow cells (FLO-PRO114M). Load 4-5 barcoded samples per flow cell.
For adaptive sampling the following expansion packs are required:
- Sequencing Auxiliary Vials V14 (EXP-AUX003)
- Flow Cell Wash Kit (EXP-WSH004)
Note
IMPORTANT: The Native Adapter (NA) used in this kit and protocol is not interchangeable with other sequencing adapters. The Native Barcode Auxiliary V14 (EXP-NBA114) expansion pack allows unused barcodes to be utilised.
Input DNA recommendations:
Quality check DNA length, quantity and purity before starting. It is important that the input DNA meets the quantity and quality requirements (see ONT notes on input DNA document).
High molecular weight DNA may require shearing prior to adaptive sampling to minimise pore blocking and increase read length N50 (see ONT notes of DNA fragmentation). We perform mechanical DNA fragmentation using Diagenode Megaruptor‱ 3 DNA shearing kit (and DNAFluid+ kit for viscous samples) to ensure uniform fragment lengths of 12-15 kb.
Shorter fragments are preferentially sequenced compared to longer fragments in a pool, which can be minimised by pooling samples of a similar size so that an equimolar mass of each sample is sequenced. Four-five barcoded libraries may be loaded per flow cell.
6 µg gDNA is enough starting material for fragmentation and two side by side library preparations per sample. This should yield enough library for 3x library loads at the optimal molar quantity of 50 fmol (see ONT notes on adaptive sampling).
Third-party reagents
Oxford Nanopore Technologies have validated and recommend the use of all the third-party reagents used in this protocol. Alternatives have not been tested.
Native Barcoding Kit 24 V14 (SQK-NBD114.24) contents
Note
ONT are in the process of reformatting their kits including reducing the concentration of EDTA (see Early Access product [rev P]).
Single-use tubes format with higher EDTA concentration:
| A | B | C | D | E | |
| Name | Acronym | Cap colour | No. of vials | Fill volume per vial (µl) | |
| Native Barcodes | NB01-24 | Clear | 24 (one per barcode) | 20 | |
| DNA Control Sample | DCS | Yellow | 2 | 35 | |
| Native Adapter | NA | Green | 1 | 40 | |
| Sequencing Buffer | SB | Red | 1 | 700 | |
| Library Beads | LIB | Pink | 1 | 600 | |
| Library Solution | LIS | White cap, pink label | 1 | 600 | |
| Elution Buffer | EB | Black | 1 | 500 | |
| AMPure XP Beads | AXP | Amber | 4 | 1,200 | |
| Long Fragment Buffer | LFB | Orange | 1 | 1,800 | |
| Short Fragment Buffer | SFB | Clear | 1 | 1,800 | |
| EDTA | EDTA | Clear | 1 | 700 | |
| Flow Cell Flush | FCF | Blue | 6 | 1,170 | |
| Flow Cell Tether | FCT | Purple | 1 | 200 |
Note
This Product Contains AMPure XP Reagent Manufactured by Beckman Coulter, Inc. and can be stored at -20°C with the kit without detriment to reagent stability.
Native barcode sequences:
| A | B | C | |
| Component | Forward sequence | Reverse sequence | |
| NB01 | CACAAAGACACCGACAACTTTCTT | AAGAAAGTTGTCGGTGTCTTTGTG | |
| NB02 | ACAGACGACTACAAACGGAATCGA | TCGATTCCGTTTGTAGTCGTCTGT | |
| NB03 | CCTGGTAACTGGGACACAAGACTC | GAGTCTTGTGTCCCAGTTACCAGG | |
| NB04 | TAGGGAAACACGATAGAATCCGAA | TTCGGATTCTATCGTGTTTCCCTA | |
| NB05 | AAGGTTACACAAACCCTGGACAAG | CTTGTCCAGGGTTTGTGTAACCTT | |
| NB06 | GACTACTTTCTGCCTTTGCGAGAA | TTCTCGCAAAGGCAGAAAGTAGTC | |
| NB07 | AAGGATTCATTCCCACGGTAACAC | GTGTTACCGTGGGAATGAATCCTT | |
| NB08 | ACGTAACTTGGTTTGTTCCCTGAA | TTCAGGGAACAAACCAAGTTACGT | |
| NB09 | AACCAAGACTCGCTGTGCCTAGTT | AACTAGGCACAGCGAGTCTTGGTT | |
| NB10 | GAGAGGACAAAGGTTTCAACGCTT | AAGCGTTGAAACCTTTGTCCTCTC | |
| NB11 | TCCATTCCCTCCGATAGATGAAAC | GTTTCATCTATCGGAGGGAATGGA | |
| NB12 | TCCGATTCTGCTTCTTTCTACCTG | CAGGTAGAAAGAAGCAGAATCGGA | |
| NB13 | AGAACGACTTCCATACTCGTGTGA | TCACACGAGTATGGAAGTCGTTCT | |
| NB14 | AACGAGTCTCTTGGGACCCATAGA | TCTATGGGTCCCAAGAGACTCGTT | |
| NB15 | AGGTCTACCTCGCTAACACCACTG | CAGTGGTGTTAGCGAGGTAGACCT | |
| NB16 | CGTCAACTGACAGTGGTTCGTACT | AGTACGAACCACTGTCAGTTGACG | |
| NB17 | ACCCTCCAGGAAAGTACCTCTGAT | ATCAGAGGTACTTTCCTGGAGGGT | |
| NB18 | CCAAACCCAACAACCTAGATAGGC | GCCTATCTAGGTTGTTGGGTTTGG | |
| NB19 | GTTCCTCGTGCAGTGTCAAGAGAT | ATCTCTTGACACTGCACGAGGAAC | |
| NB20 | TTGCGTCCTGTTACGAGAACTCAT | ATGAGTTCTCGTAACAGGACGCAA | |
| NB21 | GAGCCTCTCATTGTCCGTTCTCTA | TAGAGAACGGACAATGAGAGGCTC | |
| NB22 | ACCACTGCCATGTATCAAAGTACG | CGTACTTTGATACATGGCAGTGGT | |
| NB23 | CTTACTACCCAGTGAACCTCCTCG | CGAGGAGGTTCACTGGGTAGTAAG | |
| NB24 | GCATAGTTCTGCATGATGGGTTAG | CTAACCCATCATGCAGAACTATGC |
Designing an experiment with adaptive sampling
After considering factors like % of the genome to enrich for, read lengths and response time, the next step is to collate the input files needed for an adaptive sampling experiment. These are:
- A genomic reference: this must be a FASTA or a pre-calculated minimap2 index
- A .bed file with the genomic coordinates of the target region/s, including flanking "buffer" regions (see ONT notes on adaptive sampling)
Note that “Buffer” regions are flanking regions added to both sides of every single target described in the .bed file. These regions allow reads which begin with a sequence that does not initially map to the target region, but may extend into the target region, to be accepted. By accepting reads which map into these flanking regions, the number of accepted reads that hit our target is increased. ONT recommend setting the buffer size to the read-N10 of the library (this is point at which 10% of the bases sequenced are from reads that are this length or longer). Having a buffer size that exceeds the length of all reads present in the library will lead to reads being accepted and sequenced that never enter the target region.
Note that if you do not input a .bed file, then the entire FASTA/minimap2 index file will be used for enrichment or depletion.
PromethION 24 computer requirements for adaptive sampling
The PromethION 24 DAU device with 2 x Nvidia Quadro GV100 GPUs allows ≤3 positions/flow cells to run with real-time high accuracy basecalling required for adaptive sampling. Exceeding these recommendations may lead to an increased decision time and less enrichment.
Check your PromethION flow cell
Oxford Nanopore Technologies will replace PromethION flow cell/s with <5000 pores when the storage recommendations have been followed, if the check is performed within three months of purchase and the result is reported within two days of performing the flow cell check.
Troubleshooting
Below is a list of the most commonly encountered issues, with some suggested causes and solutions. Also consult the FAQ section available on the Nanopore Community Support section.
For adaptive sampling-specific troubleshooting, refer to the adaptive-sampling-ADS_S1016_v1_revJ_12Nov2020-any document and ONT notes on adaptive sampling.
If issues persists, contact ONT Technical Support ([email protected]) by email or use LiveChat in the Nanopore Community.
Issues during DNA/RNA extraction and library preparation for Kit 14:
Low sample quality
| A | B | C | |
| Observation | Possible cause | Comments and actions | |
| Low DNA purity (Nanodrop reading for DNA OD 260/280 is <1.8 and OD 260/230 is <2.0–2.2) | The DNA extraction method does not provide the required purity | The effects of contaminants are shown in the Contaminants document. Please try an alternative extraction method that does not result in contaminant carryover. Consider performing an additional SPRI clean-up step. |
Low DNA recovery after AMPure bead clean-up
| A | B | C | |
| Observation | Possible cause | Comments and actions | |
| Low recovery | DNA loss due to a lower than intended AMPure beads-to sample ratio | 1. AMPure beads settle quickly, so ensure they are well resuspended before adding them to the sample. 2. When the AMPure beads-to-sample ratio is lower than 0.4:1, DNA fragments of any size will be lost during the clean-up. | |
| Low recovery | DNA fragments are shorter than expected | The lower the AMPure beads-to-sample ratio, the more stringent the selection against short fragments. Please always determine the input DNA length on an agarose gel (or other gel electrophoresis methods) and then calculate the appropriate amount of AMPure beads to use. | |
| Low recovery after end-prep | The wash step used ethanol <70% | DNA will be eluted from the beads when using ethanol <70%. Make sure to use the correct percentage |
Issues during the sequencing run for Kit 14:
Fewer pores at the start of sequencing than after Flow Cell Check
| A | B | C | |
| Observation | Possible cause | Comments and actions | |
| MinKNOW reported a lower number of pores at the start of sequencing than the number reported by the Flow Cell Check | An air bubble was introduced into the nanopore array | After the Flow Cell Check it is essential to remove any air bubbles near the priming port before priming the flow cell. If not removed, the air bubble can travel to the nanopore array and irreversibly damage the nanopores that have been exposed to air. | |
| The flow cell is not correctly inserted into the device | Stop the sequencing run, remove the flow cell from the sequencing device and insert it again,checking that the flow cell is firmly seated in the device and that it has reached the target temperature. If applicable, try a different position on the device. | ||
| Contaminations in the library damaged or blocked the pores | The pore count during the Flow Cell Check is performed using the QC DNA molecules present in the flow cell storage buffer. At the start of sequencing, the library itself is used to estimate the number of active pores. Because of this, variability of about 10% in the number of pores is expected. A significantly lower pore count reported at the start of sequencing can be due to contaminants in the library that have damaged the membranes or blocked the pores. Alternative DNA extraction or purification methods may be needed to improve the purity of the input material. See the ONT notes on DNA contaminants document. |
MinKNOW script failed
| A | B | C | |
| Observation | Possible cause | Comments and actions | |
| MinKNOW shows "Scriptfailed" | Restart the computer and then restart MinKNOW. If the issue persists, please collect the MinKNOW log files and contact Technical Support. If you do not have another sequencing device available, ONT recommend storing the flow cell and the loaded library at 4°C and contact Technical Support for further storage guidance. |
Pore occupancy below 40%
| A | B | C | |
| Observation | Possible cause | Comments and actions | |
| Pore occupancy <40% | Not enough library was loaded on the flow cell | 50 fmol of good quality library needs to be loaded onto PromethION R10.4.1 flow cells for adaptive sampling. Please quantify the library before loading and calculate the molar quantity using an online weight to molar quantity calculator. | |
| Pore occupancy close to 0 | Ethanol was used instead of LFB or SFB at the wash step after sequencing adapter ligation | Ethanol can denature the motor protein on the sequencing adapters. Make sure the LFB or SFB buffer was used after ligation of sequencing adapters. | |
| Pore occupancy close to 0 | No tether on the flow cell | Tethers are adding during flow cell priming. Make sure FCT was added to FCF before priming. Be sure to use FCT+FCF using kit 14 chemistry (Native Barcoding Kit 24 V14 SQK-NBD114.24 and Sequencing Auxiliary Vials V14 EXP-AUX003). |
Shorter than expected read length
| A | B | C | |
| Observation | Possible cause | Comments and actions | |
| Shorter than expected read length | Unwanted fragmentation of DNA sample | Read length reflects input DNA fragment length. Input DNA can be fragmented during extraction and library prep. 1. Please review the Extraction Methods in the Nanopore Community for best practice for extraction. 2. Visualise the input DNA fragment length distribution on an agarose gel (or Agilent Femto Pulse) before proceeding to the library prep. In the image, Sample 1 is of high molecular weight, whereas Sample 2 has been fragmented. 3. During library prep, avoid pipetting and vortexing when mixing reagents. Flicking or inverting the tube is sufficient. |
Large proportion of unavailable pores
| A | B | C | |
| Observation | Possible cause | Comments and actions | |
| Large proportion of unavailable pores (shown as blue in the channels panel and pore activity plot). The pore activity plot above shows an increasing proportion of "unavailable" pores over time. | Contaminants are present in the sample | Some contaminants can be cleared from the pores by the unblocking function built into MinKNOW. If this is successful, the pore status will change to "sequencing pore". If the proportion of unavailable pores remains large or increases, a nuclease flush using the Flow Cell Wash Kit (EXPWSH004) can be performed. | |
| Persistent application of the “unblock” voltage reversal mechanism to reject off-target reads during adaptive sampling. | Pore becomes “unavailable” for sequencing (terminally blocked) and over time, as more pores become unavailable, the rate of data acquisition begins to slow. Due to the persistent application of the “unblock” mechanism in adaptive sampling runs (to reject off-target reads), the rate of pores becoming terminally blocked is often higher when compared with conventional runs. This effect can be exacerbated when input fragment lengths are longer. In order to maintain high data outputs with longer read libraries in adaptive sampling runs, ONT recommend performing nuclease flushes (using EXPWSH004) to clear terminally blocked pores, and then re-priming and re-loading library. |
Large proportion of inactive pores
| A | B | C | |
| Observation | Possible cause | Comments and actions | |
| Large proportion of inactive/unavailable pores (shown as light blue in the channels panel and pore activity plot. Pores or membranes are irreversibly damaged) | Air bubbles have been introduced to the flowcell | Air bubbles introduced through flow cell during priming and library loading can irreversibly damage the pores. Watch the Priming and loading your flow cell video for best practice: Nanopore Learning / Lesson library / Priming and loading your flow cell | |
| Large proportion of inactive/unavailable pores | Contaminants are present in the sample | The effects of contaminants are described in the ONT notes DNA Contaminants document. Please try an alternative extraction method that does not result in contaminant carryover. |
Reduction in sequencing speed and q-score later into the run
| A | B | C | |
| Observation | Possible cause | Comments and actions | |
| Reduction in sequencing speed and qscore later into the run | Fast fuel consumption | Add more fuel to the flow cell by re-priming the flow cell with FCF+FCT, then load library to the flow cell. |
Lower than expected sequencing depth in adaptive sampling
| A | B | C | |
| Observation | Possible cause | Comments and actions | |
| Lower than expected sequencing depth during adaptive sampling | Not enough “buffer” region in the target described in the .bed file. | Flanking “buffer” regions are added to either side of the target region of interest described in the .bed file. By accepting reads which map into these flanking regions we increase the number of accepted reads that hit our target: The size of the buffer chosen relates to the read length of the underlying library – ONT recommend setting the buffer size to the read-N10 of the library (this is point at which 10% of the bases sequenced are from reads that are this length or longer). Having a buffer size that exceeds the length of all reads present in the library will lead to reads being accepted and sequenced that never enter the target region. |
Disproportionate sequencing of individual barcoded samples within a pool
| A | B | C | |
| Observation | Possible cause | Comments and actions | |
| Disproportionate sequencing of individual barcoded samples within a pool | shorter fragments within a pool are efficiently sequenced | Ensure samples of similar fragment lengths are pooled together and run on the same flow cell. Run and carefully gate each sample on a Femto Pulse in order to appropriately pool samples together. | |
| Unequal molar quantities of samples are barcoded and pooled | Ensure that an equimolar mass of each sample is taken forward into barcoding and pooling. With adaptive sampling, barcode balancing is available as a beta feature which allows users to preferentially sequence underrepresented barcodes in their samples to balance the read data across the barcodes based on the reference file provided. Please note, with increasing the number of reads sequenced for these barcodes, the overall data output for all reads may be reduced. |
Temperature fluctuation
| A | B | C | |
| Observation | Possible cause | Comments and actions | |
| Temperature fluctuation | The flow cell has lost contact with the device | Check that there is a heat pad covering the metal plate on the back of the flow cell. Re-insert the flow cell and press it down to make sure the connector pins are firmly in contact with the device. If necessary, move the flow cell to a new position and if the problem persists, contact Technical Services. |
Failed to reach target temperature
| A | B | C | |
| Observation | Possible cause | Comments and actions | |
| MinKNOW shows "Failed to reach target temperature" | The instrument was placed in a location that is colder than normal room temperature, or a location with poor ventilation (which leads to the flow cells overheating) | MinKNOW has a default timeframe for the flow cell to reach the target temperature. Once the timeframe is exceeded, an error message will appear and the sequencing experiment will continue. However, sequencing at an incorrect temperature may lead to a decrease in throughput and lower q-scores. Please adjust the location of the sequencing device to ensure that it is placed at room temperature with good ventilation, then re-start the process in MinKNOW. |
Materials
Workflow, consumables and equipment
Notes before starting:
- Perform fragmentation on 6000 ng gDNA per sample (provides enough starting material for two side by side library preparations.
- 2x 1200-1500 ng fragmented gDNA per sample if using ≤5 barcodes
- These quantities yield 3x library loads at the optimal molar quantity of 50 fmol (see ONT notes on adaptive sampling).
- Load 4-5 barcoded libraries per flow cell.
- The PromethION 24 DAU device with 2 x Nvidia Quadro GV100 GPUs allows ≤3 positions/flow cells to run with real-time high accuracy basecalling required for adaptive sampling. Exceeding these recommendations may lead to an increased decision time and less enrichment.
- Adaptive sampling requires a genomic reference (a FASTA or a pre-calculated minimap2 index) and a .bed file with the genomic coordinates of the target region/s (see Designing and running an experiment with adaptive sampling section in 'adaptive-sampling-ADS_S1016_v1_revJ_12Nov2020-any' document)
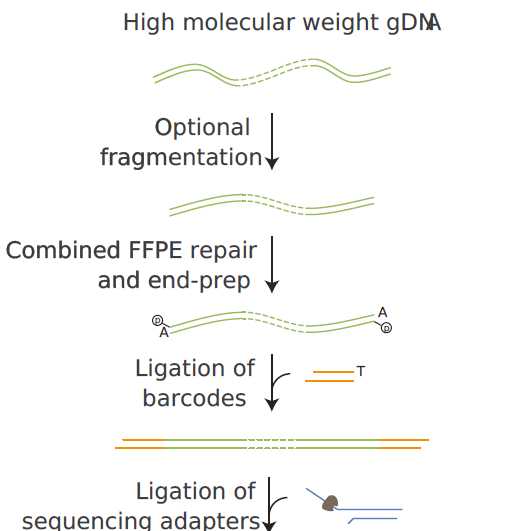
Adaptive sampling workflow:
- Optional fragmentation of 6000 ng gDNA per sample
- SPRI clean-up, volume reduction and quantitation
- FFPE repair and end-prep on 2x 1200-1500 ng gDNA per sample if using ≤5 barcodes
- Ligation of barcodes
- ligation of sequencing adapters
- Prime and load 50 fmol prepared library onto the flow cell
- Start sequencing
- 2x optional flow cell wash, re-prime and library reload
Consumables:
- Native Barcoding Kit 24 V14 (SQK-NBD114.24)
- ProNex‱ beads (Promega)
- NEBNext FFPE DNA Repair Mix (NEB, M6630)
- NEBNext Ultra II End repair/dA-tailing Module (NEB, cat # E7546)
- NEB Blunt/TA Ligase Master Mix (NEB, cat # M0367)
- NEBNext Quick Ligation Module (NEB, cat # E6056)
- 1.5 ml Eppendorf DNA LoBind tubes
- 0.2 ml thin-walled PCR tubes
- Nuclease-free water (e.g. ThermoFisher, cat # AM9937)
- Freshly prepared 80% ethanol in nuclease-free water
- Qubit™ Assay Tubes (Invitrogen, Q32856)
- Qubit dsDNA HS Assay Kit (ThermoFisher, cat # Q32851)
- PromethION R10.4.1 flow cells (FLO-PRO114M).
- Sequencing Auxiliary Vials V14 (EXP-AUX003)
- Flow Cell Wash Kit (EXP-WSH004)
Equipment:
- Hula mixer (gentle rotator mixer)
- Microfuge
- Magnetic rack
- Vortex mixer
- Thermal cycler
- Heating block for 1.5ml tubes set to 37 °C
- P1000 pipette and tips
- P200 pipette and tips
- P100 pipette and tips
- P20 pipette and tips
- P10 pipette and tips
- P2 pipette and tips
- Ice bucket with ice
- Timer
- Eppendorf 5424 centrifuge (or equivalent)
- Qubit fluorometer (or equivalent for QC check)
- PromethION 24/48 device
Optional Equipment
- Nanodrop spectrophotometer
- Agilent Femto Pulse and Genomic DNA 165 kb kit
NEB Blunt/TA Ligase Master Mix Catalog #M0367
NEBNext FFPE DNA Repair Mix - 96 rxnsNew England BiolabsCatalog #M6630L
NEBNext® Ultra™ II End Repair/dA-Tailing ModuleNew England BiolabsCatalog #E7546
NEBNext Quick Ligation ModuleNew England BiolabsCatalog #E6056S
Nuclease-Free Water (not DEPC-Treated)Thermo Fisher ScientificCatalog #AM9937
Qubit™ Assay TubesInvitrogen - Thermo FisherCatalog #Q32856
Qubit™ dsDNA HS Assay KitInvitrogen - Thermo FisherCatalog #Q32851
Prepare for your experiment
This protocol is based on ligation-sequencing-gdna-native-barcoding-v14-sqk-nbd114-24-NBE_9169_v114_revF_15Sep2022-promethion with some modifications for adaptive sampling.
Refer to the ONT notes on adaptive sampling for further information and creating a .BED file.
- Ensure you have your Native Barcoding Kit 24 V14 (SQK-NBD114.24), the correct equipment and third-party reagents.
- Check your PromethION R10.4.1 flow cell/s (FLO-PRO114M) to ensure there are >5000 pores.
Prepare samples for your experiment
- This protocol is for barcoding 4-5 samples per flow cell.
- Only 2 flow cells may be run simultaneously with high accuracy basecalling required for adaptive sampling.
- Extract DNA, and check its length, quantity and purity.
High molecular weight DNA may require shearing prior to adaptive sampling. If shearing is not required, proceed with clean-up and volume reduction
Shear 6 µg gDNA using Diagenode Megaruptor‱ 3 DNA shearing kit (and DNAFluid+ kit for viscous samples) to obtain uniform fragment lengths of 12-15 kb.
Check fragment length using Agilent Femto Pulse Genomic 165 kb kit (or similar electrophoresis method).
Note
6 µg gDNA starting material allows two side by side library preparations per sample, which usually provides 3x library loads at the optimal molar quantity of 50 fmol (see ONT notes on adaptive sampling).
Perform a 1X ProNex‱ bead clean-up on fragmented gDNA
Allow ProNex‱ beads to get to Room temperature and resuspend by vortexing.
Add 1X the volume of resuspended ProNex‱ beads to gDNA and mix by flicking the tube.
Incubate on a Hula rotator mixer for 00:10:00 at Room temperature .
10m
Meanwhile, prepare sufficient fresh 80% ethanol in nuclease-free water to completely cover the samples. Allow enough for two washes, with some excess.
Spin down the samples and pellet the beads on a magnet for 00:02:00 until the eluate is clear and colourless. Keep the tubes on the magnet, pipette off the supernatant and retain (SN1).
Note
Keep the supernatant and quantitate in the event of significant gDNA loss following elution
2m
Keep the tube on the magnet and wash the beads with enough freshly prepared 80% ethanol to cover the pellet. Remove the ethanol using a pipette and discard.
Note
Do not disturb the pellet. If the pellet was disturbed, wait for beads to pellet again before removing the ethanol.
Repeat the previous step
Briefly spin down and place the tubes back on the magnet for the beads to pellet. Pipette off and discard any residual ethanol. Do not allow the pellet to dry out.
Remove the tubes from the magnetic rack and resuspend the pellet in 25 µL nuclease-free water. Spin down and incubate for 00:10:00 at Room temperature .
Note
incubating at 37 °C with occasional flicking can improve elution efficiency
10m
Pellet the beads on a magnet for 00:02:00 until the eluate is clear and colourless.
2m
Remove and retain 25 µL of eluate into a clean 1.5 ml Eppendorf DNA LoBind tube.
Quantify 1 µL of each eluted sample using a Qubit fluorometer.
Note
In the event of significant gDNA loss, quantitate SN1 using a Qubit fluorometer, and if necessary, repeat the 1X ProNex‱ bead clean-up on SN1
If necessary, dilute gDNA with nuclease free water to obtain 2X 12 µL aliquots per sample at a concentration of 1200-1500 ng in clean 0.2 ml thin-walled PCR tubes.
Note
You may store the samples at 4 °C Overnight .
DNA repair and end-prep
- Thaw the AMPure XP Beads (AXP) at Room temperature
- Thaw NEBNext FFPE DNA Repair Buffer, Ultra II End-Prep Reaction Buffer, Ultra II End-Prep Enzyme Mix and NEBNext FFPE DNA Repair Mix On ice .
Flick and/or invert the reagent tubes to ensure they are well mixed. Spin down tubes before opening.
Note
Do not vortex the FFPE DNA Repair Mix or Ultra II End Prep Enzyme Mix.
Note
The Ultra II End Prep Buffer and FFPE DNA Repair Buffer may have a little precipitate. Allow the mixture to come to room temperature and pipette the buffer up and down several times to break up the precipitate, followed by vortexing the tube for 00:00:30 to solubilise any precipitate.
The FFPE DNA Repair Buffer may have a yellow tinge and is fine to use if yellow.
Optional addition of DNA Control Sample (DCS).
Note
- Introduction of DCS can help users distinguish between sample failure and library preparation failure.
- Dilute your DNA Control Sample (DCS) by adding 105 µL Elution Buffer (EB) directly to one DCS tube. Mix gently by pipetting and spin down.
- One tube of diluted DNA Control Sample (DCS) is enough for 140 samples. Excess can be stored at -20°C in the freezer.
- Include 1 µL DNA Control Sample (DCS) in your library prep for troubleshooting purposes.
Prepare a mastermix of the end-prep and DNA repair reagents for the total number of samples and add 3 µL to each sample tube.
| A | B | |
| Reagent | Volume | |
| DNA sample | 12 µl | |
| NEBNext FFPE DNA Repair Buffer | 0.875 µl | |
| Ultra II End-prep Reaction Buffer | 0.875 µl | |
| Ultra II End-prep Enzyme Mix | 0.75 µl | |
| NEBNext FFPE DNA Repair Mix | 0.5 µl | |
| Total | 15 µl |
Note
If 1 µL DCS is to be included, use 11 µL DNA sample
Pipette mix 10 - 20 times and spin down in a centrifuge.
Using a thermal cycler, incubate at 20 °C for 00:30:00 and 65 °C for 00:05:00 .
35m
Transfer each sample into a clean 1.5 ml Eppendorf DNA LoBind tube.
Resuspend the AMPure XP beads (AXP) by vortexing.
Add 15 µL of resuspended AMPure XP Beads (AXP) to each end-prep reaction and mix by flicking the tube.
Incubate on a Hula rotator mixer for 00:10:00 at Room temperature .
10m
Prepare sufficient fresh 80% ethanol in nuclease-free water for all of your samples. Allow enough for 400 µl per sample, with some excess.
Spin down the samples and pellet the beads on a magnet for 00:02:00 until the eluate is clear and colourless. Keep the tubes on the magnet, pipette off the supernatant and retain (SN2)
2m
Keep the tube on the magnet and wash the beads with 200 µL of freshly prepared 80% ethanol without disturbing the pellet. Remove the ethanol using a pipette and discard.
Note
If the pellet was disturbed, wait for beads to pellet again before removing the ethanol.
Repeat the previous step
Briefly spin down and place the tubes back on the magnet for the beads to pellet. Pipette off any residual ethanol. Do not allow the pellet to dry out.
Remove the tubes from the magnetic rack and resuspend the pellet in 10 µL nuclease-free water. Spin down and incubate for 00:10:00 at 37 °C .
10m
Pellet the beads on a magnet for 00:02:00 until the eluate is clear and colourless.
2m
Remove and retain 10 µL of eluate into a clean 1.5 ml Eppendorf DNA LoBind tube.
Quantify 1 µL of each eluted sample using a Qubit fluorometer.
Note
In the event of significant sample loss, quantitate SN2 using a Qubit fluorometer, and if necessary, repeat the AMPure XP Beads (AXP) bead clean-up on SN2
Take forward an equimolar mass of each sample to be barcoded into the native barcode ligation step:
Use an online weight to molar quantity calculator to identify which sample has the lowest molar concentration in 7.5 µL . Dilute other samples to the same molar quantity in 7.5 µL .
Note
Shorter fragments are preferentially sequenced compared to longer fragments in a pool. This can be minimised by pooling samples of a similar size so that an equimolar mass of each sample is sequenced in the same flow cell.
Note
You may store the samples at 4 °C Overnight .
Native barcode ligation
45m 45s
- Thaw the NEB Blunt/TA Ligase Master Mix at Room temperature , spin briefly and mix by performing 10 full volume pipette mixes. Store On ice .
- Thaw the EDTA at Room temperature and mix by vortexing. Then spin down and place On ice .
- Thaw the required number of Native Barcodes (NB01-24) at Room temperature . Individually mix the barcodes by pipetting, spin down and place them On ice .
Note
Only use one barcode per sample. Select a unique barcode for each sample to be run together on the same flow cell.
The Native Barcoding Kit 24 V14 (SQK-NBD114.24) contains 24 barcodes. Unused barcodes can be utilised with the Native Barcode Auxiliary V14 (EXP-NBA114) expansion pack.
In clean 0.2 ml PCR-tubes add the reagents in the following order:
Between each addition, pipette mix 10 - 20 times
| A | B | |
| Reagent | Volume | |
| End-prepped DNA | 7.5 µl | |
| Native Barcode (NB01-24) | 2.5 µl | |
| Blunt/TA Ligase Master Mix | 10 µl | |
| Total | 20 µl |
Thoroughly mix the reaction by gently pipetting and briefly spinning down.
Incubate for 00:20:00 at Room temperature .
20m
Add 2 µL EDTA to each reaction and mix thoroughly by pipetting and spin down briefly.
Note
EDTA is added at this step to stop the reaction.
Pool all the barcoded samples in a 1.5 ml Eppendorf DNA LoBind tube.
| A | B | C | D | |
| Volume per sample | For 4 samples | For 5 samples | ||
| Total volume | 22 µl | 88 µl | 110 µl |
Note
Check the volume after pooling to ensure all the liquid has been taken forward.
Resuspend the AMPure XP Beads (AXP) by vortexing.
Add 0.8X AMPure XP Beads (AXP) to the pooled reaction, and mix by pipetting.
| A | B | C | D | |
| Volume per sample | For 4 samples | For 5 samples | ||
| Volume of AXP | 18 µl | 72 µl | 90 µl |
Incubate on a Hula rotator mixer for 00:10:00 at Room temperature .
10m
Prepare 2 mL of fresh 80% ethanol in nuclease-free water.
Spin down the sample and pellet on a magnet for 00:05:00 . Keep the tube on the magnetic rack until the eluate is clear and colourless. Pipette off the supernatant and retain (SN3).
5m
Keep the tube on the magnetic rack and wash the beads with 700 µL of freshly prepared 80% ethanol without disturbing the pellet. Remove the ethanol using a pipette and discard.
Note
If the pellet was disturbed, wait for beads to pellet again before removing the ethanol.
Repeat the previous step
Spin down and place the tube back on the magnetic rack. Pipette off any residual ethanol. Do not dry the pellet to dry out.
Remove the tube from the magnetic rack and resuspend the pellet in 32 µL nuclease-free water by gently flicking.
Incubate for 00:10:00 at 37 °C .
Every 2 minutes, agitate the sample by gently flicking for 00:00:10 to encourage DNA elution.
10m 10s
Pellet the beads on a magnetic rack for 00:02:00 until the eluate is clear and colourless.
2m
Remove and retain 32 µL of eluate into a clean 1.5 ml Eppendorf DNA LoBind tube.
Quantify 1 µL of eluted sample using a Qubit fluorometer.
Note
In the event of significant sample loss, quantitate SN3 using a Qubit fluorometer, and if necessary, repeat the AMPure XP Beads (AXP) bead clean-up on SN3.
Note
You may store the sample at 4 °C Overnight .
Adapter ligation and clean-up
40m 45s
- Thaw NEBNext Quick Ligation Reaction Buffer (5X) at Room temperature . Spin down and ensure the reagent is fully mixed by performing 10 full volume pipette mixes. Store On ice .
Note
The NEBNext Quick Ligation Reaction Buffer (5x) may have a little precipitate. Allow the mixture to come to room temperature and pipette the buffer up and down several times to break up the precipitate, followed by vortexing the tube for several seconds to ensure the reagent is thoroughly mixed.
- Spin down the Native Adapter (NA) and Quick T4 DNA Ligase, pipette mix and place On ice .
Note
- Do NOT vortex the Quick T4 DNA Ligase.
- The Native Adapter (NA) used in this kit and protocol is not interchangeable with other sequencing adapters. The Native Barcode Auxiliary V14 (EXP-NBA114) expansion pack allows unused barcodes to be utilised.
- Thaw the Long Fragment Buffer (LFB) and Elution Buffer (EB) at Room temperature and mix by vortexing. Then spin down and place On ice .
Note
Use Long Fragment Buffer (LFB) in the clean-up step after adapter ligation to enrich for DNA fragments >3 kb.
In a 1.5 ml Eppendorf LoBind tube, mix in the following order:
Between each addition, pipette mix 10 - 20 times.
| A | B | |
| Reagent | Volume | |
| Pooled barcoded sample | 30 µl | |
| Native Adapter (NA) | 5 µl | |
| NEBNext Quick Ligation Reaction Buffer (5X) | 10 µl | |
| Quick T4 DNA Ligase | 5 µl | |
| Total | 50 µl |
Thoroughly pipette mix the reaction and briefly spin down.
Incubate the reaction for 00:20:00 at Room temperature .
20m
Resuspend the AMPure XP Beads (AXP) by vortexing.
Add 40 µL of resuspended AMPure XP Beads (AXP) to the reaction and mix by pipetting.
Incubate on a Hula rotator mixer for 00:10:00 at Room temperature .
10m
Spin down the samples and pellet the beads on a magnet for 00:02:00 until the eluate is clear and colourless. Keep the tubes on the magnet, pipette off the supernatant and retain (SN4)
2m
Wash the beads by adding 125 µL Long Fragment Buffer (LFB).
Remove from the magnet, flick the beads to resuspend, spin down, incubate for 00:02:00 at Room temperature before returning the tube to the magnetic rack.
Pellet the beads on a magnetic rack for 00:02:00 until the eluate is clear and colourless. Remove the supernatant using a pipette and discard.
Note
This clean-up step uses Long Fragment Buffer (LFB) rather than 80% ethanol to wash the beads. The use of ethanol will be detrimental to the sequencing reaction.
4m
Repeat the previous step
4m
Spin down and place the tube back on the magnet. Pipette off any residual supernatant. Do not allow the pellet to dry out.
Remove the tube from the magnetic rack and resuspend the pellet in 25 µL Elution Buffer (EB). Spin down and incubate for 00:10:00 at 37 °C . Every 2 minutes, agitate the sample by gently flicking for 00:00:10 to encourage DNA elution.
10m 10s
Pellet the beads on a magnet for 00:02:00 until the eluate is clear and colourless.
2m
Remove and retain 25 µL of eluate containing the DNA library into a clean 1.5 ml Eppendorf DNA LoBind tube.
Quantify 1 µL of eluted sample using a Qubit fluorometer.
Note
In the event of significant sample loss, quantitate SN4 using a Qubit fluorometer, and if necessary, repeat the AMPure XP Beads (AXP) bead clean-up on SN4.
Prepare the final library to 50 fmol in 32 µL of Elution Buffer.
Note
If the library yield is below the recommended input, use your entire yield and make up the remaining volume to 32 µl with Elution Buffer (EB).
If quantities allow, the library may be diluted in Elution Buffer (EB) for multiple library reloads or for splitting across multiple flow cells.
Store the library on ice or at 4°C until ready to load.
Note
Store libraries in Eppendorf DNA LoBind tubes at 4°C for short-term storage or repeated use, for example, re-loading flow cells between washes.
For single use and long-term storage of more than 3 months, store libraries at -80°C in Eppendorf DNA LoBind tubes.
Priming and loading the PromethION Flow Cell
- Remove flow cell/s from the fridge. Wait 00:20:00 before inserting into the PromethION to allow the flow cell to come to Room temperature .
Note
This kit is only compatible with R10.4.1 flow cells (FLO-PRO114M)
Note
Condensation can form on the flow cell in humid environments. Inspect the gold connector pins on the top and underside of the flow cell for condensation and wipe off with a lint-free wipe if any is observed. Ensure the heat pad (black pad) is present on the underside of the flow cell.
- Thaw the Sequencing Buffer (SB), Library Beads (LIB), Flow Cell Tether (FCT) and one tube of Flow Cell Flush (FCF) at Room temperature , mix by vortexing, spin down and store On ice .
20m
To prepare the flow cell priming mix, combine Flow Cell Tether (FCT) and Flow Cell Flush (FCF), as directed below:
Single-use tubes format:
Add 30 µL Flow Cell Tether (FCT) directly to a tube of Flow Cell Flush (FCF).
Sequencing Auxiliary Vials V14 (EXP-AUX003) format:
In a clean suitable tube for the number of flow cells, combine and mix the following reagents:
| A | B | |
| Reagent | Volume per flow cell | |
| Flow Cell Flush (FCF) | 1,170 µl | |
| Flow Cell Tether (FCT) | 30 µl | |
| Total volume | 1,200 µl |
Load the flow cell(s) into the docking ports on the PromethION 24/48.
Line up the flow cell with the connector horizontally and vertically before smoothly inserting into position.
Press down firmly onto the flow cell and ensure the latch engages and clicks into place.
Note
Insertion of the flow cells at the wrong angle can cause damage to the pins on the PromethION and affect your sequencing results. If you find the pins on a PromethION position are damaged, please contact [email protected] for assistance.
Turn the valve clockwise to expose the inlet port.
After opening the inlet port, draw back a small volume to remove any air bubbles:
Set a P1000 pipette tip to 200 µL .
Insert the tip into the inlet port.
Turn the wheel until the dial shows 220-230 µl, or until you see a small volume of buffer entering the pipette tip.
Note
Take care when drawing back buffer from the flow cell. Do not remove more than 20-30 µl, and make sure that the array of pores are covered by buffer at all times. Introducing air bubbles into the array can irreversibly damage pores.
Load 500 µL of the priming mix into the flow cell via the inlet port, avoiding the introduction of air bubbles. Wait 00:05:00 .
During this time, prepare the library for loading
5m
Note
The Library Beads (LIB) tube contains a suspension of beads that settle very quickly. It is vital that they are mixed thoroughly by pipetting immediately before use.
In a new 1.5 ml Eppendorf DNA LoBind tube, prepare the library for loading as follows:
| A | B | |
| Reagent | Volume per flow cell | |
| Sequencing Buffer (SB) | 100 µl | |
| Library Beads (LIB) thoroughly mixed before use | 68 µl | |
| DNA library | 32 µl | |
| Total | 200 µl |
Note
Do not vortex prepared library
Complete the flow cell priming by slowly loading 500 µL of the priming mix into the inlet port.
Wait 00:05:00 before loading library
5m
Mix the prepared library gently by pipetting up and down just prior to loading.
Using a P1000, insert the pipette tip into the inlet port and load 200 µL of library.
Close the valve to seal the inlet port.
Install the light shield on your flow cell as follows:
- Align the inlet port cut out of the light shield with the inlet port cover on the flow cell. The leading edge of the light shield should sit above the flow cell ID. Firmly press the light shield around the inlet port cover. The inlet port clip will click into place underneath the inlet port cover.
Note
For optimal sequencing output ONT recommend leaving the light shield on the flow cell when library is loaded, including during any washing and reloading steps.
- Close the PromethION lid when ready to start a sequencing run on MinKNOW.
Wait a minimum of 00:30:00 after loading the flow cells with library before initiating sequencing to increase the sequencing output.
30m
Data acquisition and high accuracy basecalling for adaptive sampling
Select 'Start Sequencing' on the start page of the MinKNOW software to set up the sequencing parameters for the experiment:
Type in the experiment name appended with a run number for the chosen flow cell/s. Label sample ID's.
Note
If sequencing needs to be stopped and the flow cell/s moved to another position, append experiment name with the next run number.
Select the Native Barcoding Kit 24 V14 (SQK-NBD114.24) used to prepare the library.
Set up the run options:
- run limit: 96 hours
- Minimum read length: 200 bp
- Adaptive sampling: select 'Enrich or deplete sequences'.
Upload an alignment reference (e.g. hg38 FASTA file) and specific sequence coordinates of interest (e.g. SCA4_human_hg38.bed).
Select 'enrich alignment matches'.
Turn 'barcode balancing' off.
Note
There is beta support to allow users to preferentially sequence underrepresented barcodes in their samples to balance the read data across the barcodes based on the reference file provided. Under this option, there will be options to balance all barcodes detected or to choose barcodes to balance. Note that with increasing the number of reads sequenced for these barcodes, the overall data output for all reads may be reduced.
'Active channel selection' is on by default.
Note
This option maximises the number of pores sequencing at the start of the experiment. If a pore is in the 'Saturated' or 'Multiple' state, the software instantly switches to a new pore in the group. If a pore is 'Recovering', MinKNOW will attempt to revert the pore back to 'Pore' or 'Sequencing' for ~5 minutes, after which it will select a new pore in the group.
By default, this option is on but can be switched off.
The time between pore scans is 1.5 hours by default.
Turn 'Reserved pore' option off to front-load sequence data acquisition.
Note
The reserve pore feature prioritises consistency and accuracy over immediacy by reserving wells where voltages have dropped until later in the run, such that other wells can catch up. Switch off this feature to fully front-load sequence data acquisition.
'Basecalling' is on by default. Click "Edit options" to specify the basecall model as 'High accuracy Basecalling'.
'Modified bases' is off by default.
Note
This can be switched on to use the CpG context models and basecall 5mC. Note that this requires additional CPU, which could slow down the high accuracy real-time basecalling required for adaptive sampling, increasing decision time and ultimately resulting in less enrichment.
Modified basecalling may be performed post-sequencing
'Barcoding' is on by default.
Note
These options are only available when a barcoding sequencing kit or expansion has been selected and automatically demultiplexes each barcode. Barcoding can be switched off and performed post-sequencing.
Click 'Edit options' to specify the barcoding options: barcode trimming, mid-read barcoding filtering and barcoding minimum score are off by default.
'Alignment' is optional.
Note
To use alignment during sequencing, upload an alignment reference file as a .fasta or .mmi file. A reference file can contain multiple entries in the same file (e.g. multiple chromosomes). Alignment hits from these files are used to populate the alignment graphs which can be viewed on the GUI.
A BED file can also be uploaded alongside the reference when there is a specific interest in a particular region of the reference (e.g. specific gene in a chromosome). Alignment hits from BED files will be highlighted in the sequencing .txt file generated in the data folder. Click 'Edit options' to open a dialogue box to upload a BED file.
Select 'output location'.
Note
ONT recommend saving your alignment files in a folder with the prefix/data or the default location MinKNOW saves your reads.
E.g. Windows: C:\data\
Mac: /Library/MinKNOW/data/
Ubuntu: /var/lib/MinKNOW/data/
For adaptive sampling experiments, there is an additional adaptive_sampling.csv file that is saved in other_reports in the run folder, which can be used for troubleshooting.
Select 'output format':
- Raw reads default is .POD5
- Basecalled reads default is .FASTQ
- Aligned reads (if selected) default is .BAM
Note
Raw reads can be saved as .POD5 or .FAST5. Note that for Kit 14 chemistry, .POD5 is the default file output, which is a Nanopore-developed file format that writes data faster, uses less compute and has smaller raw data file size.
'Filtering' is at Q9 by default for the high accuracy basecalling.
Click 'Start' to review all run options selected.
- Edits can be made by selecting the 'Edit' button.
- Select 'Advanced run options' to view the extra options selected.
- Click 'Start' to begin sequencing
Click on the flow cell to check the number of active pores.
Note
The first pore scan should report a similar number of active pores (within 10-15%) to that reported in the flow cell check. If there is a significant reduction in active pores in the first pore scan:
- restart MinKNOW
- move the flow cell to a new position in the PromethION
- reboot the host computer.
If sequencing needs to be stopped and the flow cell/s moved to another position, append experiment name with the next run number.
Monitor translocation speed, quality score and data output during the sequencing run.
If translocation speed ≤350 bps, quality score begins to drop toward Q9 and there is little increase in data output:
- pause sequencing
- Perform a flow cell wash
- Prime flow cell
- load 50 fmol prepared library
- restart sequencing
Performing a flow cell wash
- Place the tube of Wash Mix (WMX) on ice. Do not vortex.
- Thaw one tube of Wash Diluent (DIL) at room temperature. Mix the contents of Wash Diluent (DIL) thoroughly by vortexing, then spin down briefly and place on ice.
In a clean 1.5 ml Eppendorf DNA LoBind tube, prepare the following Flow Cell Wash Mix:
| A | B | |
| Reagent | Volume per flow cell | |
| Wash Mix (WMX) | 2 µl | |
| Wash Diluent (DIL) | 398 µl | |
| Total volume | 400 µl |
Mix well by pipetting and place on ice. Do not vortex.
Pause the sequencing run.
Ensure the inlet port is closed and remove any waste fluid from the waste port labelled 2 on the flow cell.
Open the inlet port and draw back a small volume to remove any air bubbles
Follow steps 62-63
Load 400 µL of the prepared Flow Cell Wash Mix into the flow cell via the inlet port, avoiding the introduction of air bubbles. Wait 01:00:00 .
1h
Prepare Priming Mix and library, load flow cell.
Follow steps 62-70
Note
It may be necessary to mop up any excess fluid that escapes from the flow cell waste ports 2 and 3 using tissue paper.
Ensure the inlet port is closed and remove any waste fluid from the waste port labelled 2 on the flow cell.
1h
Restart sequencing.
Note
Restoration of sequencing pores will be observed after a new pore scan has been performed.
Monitor translocation speed, quality score and data output during the sequencing run.
If translocation speed ≤350 bps, quality score begins to drop toward Q9 and there is little increase in data output perform a flow cell wash, re-prime the flow cell, load 50 fmol prepared library and restart sequencing steps 74-81
Ending the experiment
Flow cells are unlikely to be re-useable given the nature of adaptive sampling. However, if you would like to reuse the flow cell, follow the Flow Cell Wash steps follow steps 74-79 . Store the washed flow cell at 2 °C -8 °C .
Alternatively, flush the flow cell/s with deionised water and store ready to send back to Oxford Nanopore.
Data Analysis
Data analysis pipeline is available through https://github.com/egustavsson/long-read_SV_calling.git
Protocol references