Apr 16, 2026
Nanopore long-read metabarcoding for the comprehensive characterisation of parasitic nematodes in humans and animals: a development and diagnostic validation study
- 1University Of Melbourne



Protocol Citation: Lucas Huggins 2026. Nanopore long-read metabarcoding for the comprehensive characterisation of parasitic nematodes in humans and animals: a development and diagnostic validation study. protocols.io https://dx.doi.org/10.17504/protocols.io.dm6gpmq1jgzp/v1
Manuscript citation:
Evaluation of nanopore long-read metabarcoding for the comprehensive characterisation of parasitic nematode infections in humans and animals: a methodological development, validation and diagnostic accuracy study
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: October 19, 2025
Last Modified: April 16, 2026
Protocol Integer ID: 230237
Keywords: parasitic nematodes in human, parasitic nematode clade, parasitic nematodes of vertebrate, parasitic nematode, simultaneous characterisation of all parasitic nematode, genes for nematode species, important nematodes in the genera strongyloide, different genera across all parasitic nematode clade, nematode species, important nematode, nemabiome assay, parasitic species, emerging parasitic threat, novel nemabiome method, parasite community, unidentified parasite, greater diversity of parasitic species, parasite, parasite control program, additional host species, parasitic threat, nemabiome, oxford nanopore technologies platform, capabilities of oxford nanopore technologies platform, nanopore, unprecedented taxonomic resolution, large biobank, genera strongyloide, cryptic species, advanced diagnostic tool, significant dearth of advanced diagnostic tool, large biobank of positive control sample, animal reservoir, read sequencing
Funders Acknowledgements:
National Health and Medical Research Council
Abstract
Background
Parasitic nematodes of vertebrates impose a substantial burden on human and animal health, however there is a significant dearth of advanced diagnostic tools for their accurate characterisation. One promising but nascent approach is metabarcoding, which enables the simultaneous characterisation of all parasitic nematodes from a sample – termed the “nemabiome”. However, earlier iterations of this approach could not detect socioeconomically important nematodes in the genera Strongyloides and Trichuris. Therefore, we aimed to develop a novel nemabiome method capable of detecting all parasitic nematodes to support their accurate control in people and animals.
Methods
We employed a comparative molecular target approach to identify optimal barcoding genes for a novel method capable of detecting all parasitic nematodes affecting vertebrates. This assay leverages the long-read sequencing capabilities of Oxford Nanopore Technologies platform, to achieve unprecedented taxonomic resolution. The assay was validated using a large biobank of positive control samples (n = 154) and field samples from humans and animals (n = 190). Its diagnostic performance was benchmarked against well-established quantitative PCR (qPCR) assays.
Findings
We identified suitable barcoding genes for nematode species in clades I and IV (18S ribosomal DNA) and clades III and V (internal transcribed spacers). Validation using positive controls confirmed the assay’s ability to detect parasites representing 24 different genera across all parasitic nematode clades. Benchmarking using field samples demonstrated diagnostic performance comparable to that of qPCR (diagnostic specificity: 99.6%; diagnostic sensitivity: 86.0%), while revealing a greater diversity of parasitic species (n = 10) than conventional molecular techniques.
Interpretation
Our nemabiome assay provides a comprehensive tool for the precise characterisation of parasite communities (coinfections) affecting humans and vertebrates. Unlike target-specific molecular methods, e.g., qPCR, our approach facilitates the detection of previously unidentified parasites, including zoonotic and cryptic species, whilst also elucidating inter-host transmission pathways and animal reservoirs. Continued employment of this advanced methodology in additional host species and locations will further demonstrate its capacity to strengthen parasite control programs including tackling emerging and re-emerging parasitic threats.
Materials
- QIAamp PowerFecal Pro DNA Kit (Qiagen, Hilden, Germany)
- FastPrep24 5G instrument (MP Biomedicals, California, USA)
- Qubit™ 4 Fluorometer (Thermo Fisher Scientific, Massachusetts, USA)
- dsDNA HS assay kit
- PCR Barcoding Expansion 1-96 (EXP-PBC096)
- Ligation Sequencing Kit (SQK-LSK114)
- MinION Mk1B or PromethION 2 (P2) solo sequencers (Oxford Nanopore Technologies, Oxford, UK)
- LongAmp® Hot Start Taq 2× Master Mix (New England Biolabs, Massachusetts, USA)
- Ambion nuclease-free water (Life Technologies, California, USA)
- NucleoMag NGS Clean-up and Size Select Beads (Macherey-Nagel, Duren, Germany)
- HulaMixer (Thermo Fisher Scientific)
- T100™ Thermal Cycler (Bio-Rad, California, USA)
- 4200 TapeStation System (Agilent Technologies, California, USA)
- NEBNext® Companion Module for Oxford Nanopore Technologies® Ligation Sequencing (New England Biolabs)
- Integrated DNA Technologies (Iowa, USA) synthetic DNA constructs (positive controls).
- EXP-WSH004 Flow Cell Wash Kit (Oxford Nanopore Technologies)
- MinION or P2 solo device
- Sequencing Computer
Sampling and DNA extraction
If faecal samples were preserved in a preservative e.g. 70% ethanol, 5% w/v potassium dichromate or RNAlater then the preservative was removed and Milli-Q water was added to rehydrate the sample (rehydration for a minimum of 1-hour required).
After rehydration the samples were spun down, water removed, and the faecal sample homogenised. Next 200 mg of homogenate was transferred to a bead-beating tube for DNA extraction using a QIAamp PowerFecal Pro DNA Kit (Qiagen, Hilden, Germany). Extraction was performed as per the manufacturer’s instructions with the addition of a minor modification incorporating an extra bead-beating step by adding 800 µl of CD1 buffer to the bead-beating tube in a FastPrep24 5G instrument (MP Biomedicals, California, USA). The final elution was conducted in 100 µL of elution buffer and stored at −20°C. Extractions can also be conducted using a different DNA extraction kit appropriate for removing faecal PCR inhibitors.
All DNA extracts were quantified using a Qubit™ 4 Fluorometer (Thermo Fisher Scientific, Massachusetts, USA) using the dsDNA HS assay kit.
Nemabiome assay library preparation and nanopore sequencing
The PCR Barcoding Expansion 1-96 (EXP-PBC096) with ONT® Ligation Sequencing Kit (SQK-LSK114) was utilised to conduct the library preparation for our nemabiome assay on either the MinION Mk1B or PromethION 2 (P2) solo sequencers (Oxford Nanopore Technologies, Oxford, UK). The protocol followed was ‘Ligation sequencing amplicons - PCR barcoding (SQK-LSK114 with EXP-PBC096)’ version: PBBC9182v114rev107Mar2023, with some modifications to improve yield. Separate and different physical laboratory areas were utilised for DNA extraction, pre-PCR and post-PCR experiments with all first-step PCRs prepared in a PCR hood under sterile conditions with filter tips, following UV sterilisation of the workspace.
For the first step PCR amplification, two separate PCRs were conducted, one targeting the 18S rDNA of nematode clades I and IV and another targeting the ITS regions of the clade III and V nematodes. Both reactions were 25 µl PCRs conducted using 12.5 µl of LongAmp® Hot Start Taq 2× Master Mix (New England Biolabs, Massachusetts, USA) 8.5 µl Ambion nuclease-free water (Life Technologies, California, USA) herein referred to as water, 1 µl of forward primer, 1 µl of reverse primer and 2 µl of genomic faecal extracted DNA. The primers required include the addition of ONT adapter sequences that permit the addition of ONT indexes in a subsequent secondary PCR reaction. Hence, the primers for the 18S rDNA reaction were Nemabio18S-F_ONT: 5’-TTTCTGTTGGTGCTGATATTGCCGGTAGCAGTAACCGCCC -3’ and Nemabio18S-R_ONT: 5’-ACTTGCCTGTCGCTCTATCTTCATCATAATGATCTCTCCGCAGGTTCCACTCAC -3’, whilst for the ITS region reaction they were NemabioITS-F_ONT: 5’-TTTCTGTTGGTGCTGATATTGCCGTAGGTGAACTCGGGGAAGGATCATT -3’ and NemabioITS-R_ONT: 5’-ACTTGCCTGTCGCTCTATCTTCGCGGTAATACGAGCTGAGC -3’.
PCRs were then conducted on a T100™ Thermal Cycler (Bio-Rad, California, USA) using the following conditions for the 18S rDNA reaction: 1 cycle of 94 °C for 1 min, 24 cycles of 94 °C for 30 s, 58 °C for 30 s and 65 °C for 45 s, with a final extension of 65 °C for 10 min. For the ITS region reaction conditions were: 1 cycle of 94 °C for 1 min, 25 cycles of 94 °C for 30 s, 58 °C for 30 s and 65 °C for 55 s, with a final extension of 65 °C for 10 min. The post-PCR 25 µl product of the ITS region reaction was then added to the product from the 18S rDNA reaction to create a total reaction output of 50 µl that was taken forward for further processing.
PCR product was then added to a 96-well plate and cleaned using a 1× ratio of NucleoMag NGS Clean-up and Size Select Beads (Macherey-Nagel, Duren, Germany) with a 15 min incubation on a HulaMixer (Thermo Fisher Scientific) and two washes with freshly made 75% ethanol, followed by a final elution in 27 µl of water. Next, second step PCRs were conducted to add ONT barcodes to each samples’ amplicon and thereby permit multiplexing of up to 96 samples onto a flow cell. These secondary PCRs were 50 µl reactions utilising 25 µl of LongAmp® Taq 2× Master Mix (New England Biolabs), 24 µl of cleaned PCR product from the first PCR reaction and 1 µl of a unique barcode from the ONT PCR Barcoding Expansion 1-96 kit. Thermocycling conditions for this second reaction were 1 cycle of 95 °C for 3 min, 12 cycles of 95 °C for 15 s, 62 °C for 15 s and 65 °C for 1 min 5 s, with a final extension of 65 °C for 5 min. Secondary PCRs were followed by another clean-up step using a 0.55× ratio of NucleoMag beads to exclude and remove low molecular weight (smaller than 450 bp) PCR products, hence 27.5 µl of beads were used to clean 50 µl of PCR product with the same incubation and ethanol wash steps used as previously described and elution in 25 µl of water.
Next, correct amplification of the expected product was assessed using a subset of samples on a 4200 TapeStation System (Agilent Technologies, California, USA) and the final DNA concentrations of this subset analysed using a Qubit™ 4 Fluorometer. Subsequently, 4 µl of each barcoded and cleaned PCR product were pooled together and concentrated down using a 2.5× ratio of NucleoMag beads, washed twice with 75% ethanol and eluted in 50 µl of water. This amplicon pool was then quantified on a Qubit™ 4 Fluorometer to ensure there was adequate DNA, i.e., a minimum requirement of 1,000 ng, to be taken forward.
Final library preparation steps included DNA repair and end-prep, adapter ligation, clean-up and flow cell priming and loading was conducted exactly as described in the aforementioned ONT® protocol, making use of the NEBNext® Companion Module for Oxford Nanopore Technologies® Ligation Sequencing (New England Biolabs) and the ONT® Ligation Sequencing Kit (SQK-LSK114). The final concentration of sequencing library was always diluted to 50 fmol using an average product fragment size of 1,400 bp. 50 fmol was chosen as it is at the top end of the 35 – 50 fmol range recommended by ONT protocols for loading onto R10.4-1 flow cells.
Batches were run with no template PCR negative controls, i.e., water, and sequencing-run positive controls that were comprised of two uniquely identifiable gBlock synthetic DNA constructs (Integrated DNA Technologies, Iowa, USA) of the 16S rDNA from the bacterial species Aliivibrio fischeri and Methanopyrus kandleri. The species M. kandleri was used as a sequencing-run positive control for the 18S rDNA reaction and A. fischeri for the ITS region reaction. These sequencing-run positive control gBlocks consisted of the relevant 16S rDNA sequence flanked by the appropriate primer binding regions as two separate artificial gene constructs, the design of which, can be seen in the proceeding section.
For the accurate determination of whether field samples were truly positive for a given nematode parasite the identification of read count thresholds was critical to determine the accurate infection status of a sample. Such thresholds were calculated using a previously developed method. In brief, reads of the uniquely identifiable M. kandleri (18S rDNA target) and A. fischeri (ITS regions target) sequencing-run positive control sequences that were found in samples other than the positive control were used to inform the read cut-off threshold for a given library. Identification of these sequencing-run positive control sequences in non-positive control samples can occur due to occasional barcode index misreading, sequencing error, chimeric reads, or infrequent cross-contamination during NGS library preparation across a 96-well plate. Sequencing-run positive controls were used as a pure gBlock sequence at a concentration of ~1 × 10^6 ng/µl, that produced a post-PCR DNA concentration substantially higher than that achieved by biological samples, making these sequences pose the greatest risk of generating index misreading or cross-contamination. Therefore, to be able to take the strictest possible read cut-off threshold for a given sequencing batch, the cut-off was taken to be the highest read-count of a sequencing-run positive control sequence, i.e. M. kandleri or A. fischeri sequence, in a non-positive control sample. If parasitic nematode reads from a faecal sample were found to be lower than the batch’s threshold then the sample was defined as negative for that pathogen, i.e., a false positive.
If flow cells are to be re-used they must be DNAse-treated using the EXP-WSH004 Flow Cell Wash Kit (Oxford Nanopore Technologies), to remove the possibility of DNA contamination and carry-over from prior sequencing runs. In general we do not recommend the re-use of flow cells for the nemabiome assay as DNA carry-over from previous runs can increase the chances of false positives in subsequent runs.
Nanopore sequencing was conducted on either a MinION Mk1B device using a Legion 7i Gen 6 laptop (Lenovo, Quarry Bay, Hong Kong) that utilises a NVIDIA® GeForce RTX 3070 GPU or on a P2 solo device attached to a custom desktop computer running an Intel Core i9 13900F CPU and GeForce RTX 4070 GPU. Sequencing was initiated through MinKNOW version 24.02.8 with fast base-calling for between 22 - 47 hrs depending on the amount of data required. Sequencing time should be chosen based on achieving an average per sample sequencing depth of ~230,000 reads that pass quality filtering, i.e. the ONT fast base-calling default Q-score of 8 or greater.
Once sequencing was stopped, POD5 reads were base-called using the super accuracy base-calling model with barcodes, flanking sequences and primer sequences removed using Dorado version 7.3.11. Upon sequencing completion, the success of the sequencing run was assessed using MinKNOW to ensure reads were of the expected size and pore activity was healthy.
DNA sequences of the two sequencing-run positive control gBlock constructs used for the nemabiome assay
The gBlock sequencing-run positive control DNA sequence construct (793 bp) for the 18S ribosomal DNA (rDNA) reaction is comprised of the primer binding sites (underlined) and a region of the 16S ribosomal RNA gene of Methanopyrus kandleri (NCBI accession number AB301476.1).
5’-GGCATCAGATACCGCCGCCCCGCGGCTTAAACCCGGGAGTCGCGGGGAAACTGCGGGGATCTTGGGA CCCGGGAAGGCCGGGAGATCCCCCGGGGGTAGGGGGGTAATTCCTGTACCCCGGGGGGACCCCGAG TGGGAAGGGGCGTCCGGGTGAAACGGGTCCGACGGGTCCGAGGGGGAAACCGGGGGGAGACCCGGG ATTAGATACCGGGGTAGCTTCCGGGGTGTAACGATCGGGGATACGGTGTGGGGCGGCATGAGCCGC CCGAGGCGTCGAGGGAAGCCTTTAACTCCCGGGGATGCGGCGGATACGCGCCGCAAGCGGTAACCTT AAGGAATTGGGGGGGGAGACCACACACACCGGTGGGACGCTGGGTTTAATTGGGATTCAACCGCGGAA ACCTTACCGGGGGGCAACACGAGGATGAAGGCCAGGTTGAGCCTTGGCGGCCGACGAGCTGAGAGGA GGTCGACGGGCCCGTACGCTGGTCCGTGAGGGTGTCCTGTTAAAGTGAGGTAACAGGAGCGGAACCCC GCCCGCGGATTCCGACGGGGGCCCCCCTAAAGGGGCGCCCGGGGCCACTCTGCGGGGATCTTCCGGGTTAA GCGGCGGAAATAGTGCGGGGACGCGGAGGTCCGTTACGCCCGGAAACCCCCGCAAAACCCCGGGGCT CCAATGGCGGGGACAATAGGATCCGACCCCGGAAAGGGGGGAGAAATCCCGCTAAACCCCGGTGTAG TTCGGATTTGGGCGGTGCAACATCCCGCCCGGATAAAGGGTGGAATCGTGTAAGCTGAGGTGAACCTCGG GAGGATCATT-3’
The gBlock sequencing-run positive control DNA sequence construct (945 bp) for the internal transcribed spacer (ITS) regions reaction is comprised of the primer binding sites (underlined) and a region of the 16S ribosomal RNA gene of Aliivibrio fischeri (NCBI accession number NR_029255.1). Both constructs were synthesised by Integrated DNA Technologies (Iowa, USA).
5’-GTAGGTGAACCTGCGGAAGGACATTATTTAGAACGTGCGGCGGACGCCCTAACAACATCGCAATTCGAGC GGAACAGACTTAACCTTGAACCTTCGGGGAAGGATTGAAGGGGCTGAGGCCGGGCGGACGGGTGAGATAG CCTGGGAATTACCTTATGGTGTGGGGGATAACATTTGGAAGACGATAGCTAAACCGCGAATAATGTCTTCCG CACCGAAGAGGGGGACCTTTCGGGGCCTCCGGCCTGAAGATTAGCCCGGATGGAATTACGGTAGTTGGT GAGGTAAGAGCTCACACAGGCGGACGACTTCTCAGCTCGGTCTGAAGAGGATGACAGCAACTGGAAC TGAGACACGGTCCGACATCCTCAGGGAGGCGGACGACGGTGGGGAATATTGACAAATGGCGGAAAACCT GATGCACACCTCGGCGTGTAAGAAGGGCTCCTCGGGTGTTAAAAGATCTTTCGATGAGGGAGGAAG GTGTTGTGATTTAAATGCTGACGCATTTTGACGTTTACCTTACGAAGAAGACCCCGGCTACATCCGGTGCCA GACGCCGGGTAATAACGAGGGTGCGAGGTTCAAACTACGGGAATACCTGGGCGAAACGCCATCGAGGT GGTTCTCAAGTGAGGGGGGTGAATTTACCGGGTGACGGTGGAAGTCGGAATCGGTAAGGAATACT GAGATGCTGTGAGAGGGGTGGAATTTCAACGGTGTAAGGCTGGAATCGGTAAGGTAACGTTGAAGGAATAA CCGATGGGGAACGCGGCCCCCCTGGGAACACGACTGACACTGACATCGGAAAGCGCTGGGGGACAAAC GGATTAAGATCCCTCGGTTGATACCCCGGTAACGTTGCTGAAGAGGCTGGAAGGTAACGTTGAAGGAATC GGCTTCGGAACGTGTAAGTAAGACCCGCTGGGGACGGTGCCGAAGATTAACCTCAAAT GGCTTCGGAACGTGTAAGTAAGACCCGCTGGGGACGGTGCCGAAGATTAACCTCAAAT-3’
Nematobiome assay bioinformatic analysis
Sequencing data was demultiplexed using MinKNOW and fastq files for each barcode concatenated prior to downstream analysis. Because both the 18S rDNA and ITS region sequences were pooled and given the same barcode for each sample analysed these firstly had to be separated using pblat (https://github.com/icebert/pblat) and seqtk (https://github.com/lh3/seqtk). To conduct separation and binning of 18S rDNA and ITS sequences into different files, a database of nematode 18S rDNA sequences was built just using the region of the 18S rDNA targeted by our primers. Using a pblat minimum score value of 50, the 18S rDNA sequences from the data for each barcode were compared to our pblat 18S rDNA database and extracted to form an 18S rDNA sequence file, whilst the remaining sequences were used to form an ITS region sequence file. Script and database for sequence separation can be found at https://figshare.unimelb.edu.au/projects/Huggins_Nemabiome/231149.
The NanoCLUST bioinformatic pipeline was used as it corrects for the error rate of the utilised ONT R10.4-1 flow cells by construction of accurate consensus sequences. Overall, the pipeline conducts multiple quality control, read clustering, polishing and consensus forming steps followed by classification of consensus sequences using blastn against a database of the user’s choice. NanoCLUST has already demonstrated great accuracy, i.e. consistently >99.5% identical to reference sequences, when generating 16S rDNA, 18S rDNA and cytochrome C oxidase (COI) gene consensus sequences for bacterial, protozoan and filarial worm metabarcoding investigations.
Optimal NanoCLUST parameters for the 18S rDNA reaction were found to be to be a minimum read length of 600 bp, maximum read length of 1,200 bp, minimum cluster size of 30, HDBSCAN cluster selection epsilon value of 1, polishing with 100 reads and 300,000 reads used for UMAP projection. For the ITS region reaction optimal parameters were a minimum read length of 600 bp, maximum read length of 1,800 bp, minimum cluster size of 30, HDBSCAN cluster selection epsilon value of 1, polishing with 100 reads and 300,000 reads used for UMAP projection.
Read counts and classifications produced by NanoCLUST were taken as the final dataset generated by the nanopore sequencing assay to which other methods were compared. If NanoCLUST classification identity scores were low, i.e. <97%, then the sequence was further validated by conducting blastn against the full GenBank database. Query cover and percent identity scores returned by classification against GenBank could then be used to improve the original classification. This was only required for off-target and non-nematode sequences which were not present in the bespoke databases used by NanoCLUST and therefore could not be classified accurately by this pipeline alone. If the sequence could still not be correctly classified when compared to GenBank, then it was removed from the overarching dataset.
To ensure that consensus sequences generated by NanoCLUST were accurately classified two bespoke databases were built, one for the 18S rDNA reads and one for the ITS regions read data. For the ITS regions database all Nematoda (txid6231) rDNA sequences, i.e. those containing 18S rDNA, ITS1, 5.8S, ITS2 and 28S rDNA regions that were >200 bp and <10,000 bp and were verified and published were downloaded from NCBI’s GenBank. The script for blastn database construction and GenBank search terms used for reference sequence download can be found at https://figshare.unimelb.edu.au/projects/Huggins_Nemabiome/231149.
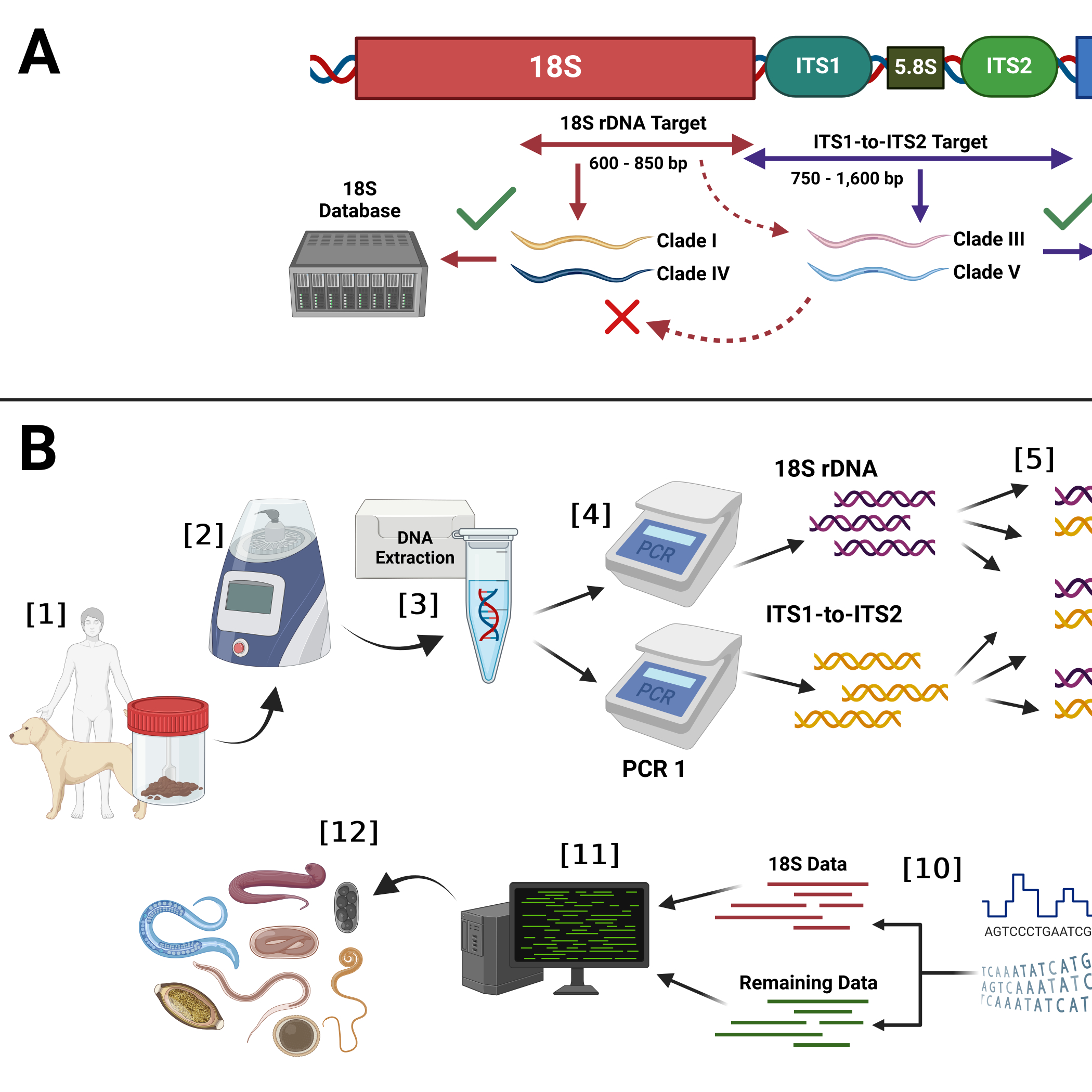
For the 18S rDNA database only relevant sequences from key clade I and IV parasitic nematode groups were downloaded and only if they were >200 bp and <10,000 bp and were verified and published. Because the 18S rDNA locus is taxonomically uninformative for clade III and V parasitic nematodes, sequences from these clades were also added to the database and annotated as ‘unidentified nematode’. This was done to ensure a user would not incorrectly use the 18S rDNA targeting component of the assay to classify clade III and V nematodes. With these amended annotations the 18S rDNA database would subsequently output taxa from clade III and V nematodes with an ‘unidentified’ classification, thereby guaranteeing that a spurious or inaccurate classification was not generated by this database. The overarching structure of rDNA targets used by the assay, nematode clades targeted by each primer pair and databases used to classify amplicons from each pair are shown in the protocol's Figure.
For both databases, if important parasitic nematode taxa were not represented due to them not being marked as published in NCBI, then verified but unpublished sequences were downloaded and incorporated. Additionally, the inclusion of both sequencing-run positive control sequences for _A. fischeri_ and _M. kandleri_ as well as important off-target species rDNA sequences were added due to the nemabiome assay occasionally co-amplifying non-nematode species such as fungi, mites, plants, platyhelminths and cestodes. Both databases are downloadable from https://figshare.unimelb.edu.au/projects/Huggins_Nemabiome/231149.