Sep 03, 2025
Version 2
iTP-Seq: a scalable profiling method to study context-dependent translational events in vitro V.2
- Mélanie illard1,
- Thibaud T. Renault1,
- C. Axel Innis1
- 1Univ. Bordeaux, Centre National de la Recherche Scientifique, Institut National de la Santé et de la Recherche Médicale, ARNA, UMR 5320, U1212, 33000 Bordeaux, France
- Mélanie illard: https://orcid.org/0000-0002-1974-8030
- C. Axel Innis: https://orcid.org/0000-0002-1974-8030



Protocol Citation: Mélanie illard, Thibaud T. Renault, C. Axel Innis 2025. iTP-Seq: a scalable profiling method to study context-dependent translational events in vitro
. protocols.io https://dx.doi.org/10.17504/protocols.io.bp2l6dqekvqe/v2Version created by Mélanie Gillard
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: August 01, 2025
Last Modified: September 03, 2025
Protocol Integer ID: 223879
Keywords: Bacterial ribosome, Antibiotics, Context-dependent in vitro translation, Next-generation sequencing, iTP-Seq, ribosome profiling, context dependence of ribosome, ribosome, short mrna footprint, mrna context, protected mrna fragment, mrna fragment, mrna sequence, mrna biotinylation, stalled ribosome, sequencing library, wide information on protein synthesis, generation sequencing, targeting antibiotic, sequenced genome, genome, reverse transcription, transcription, focused transcript library, protein synthesis, translated sequence, nascent amino acid sequence, ribo, regulation of the proteome, dna library amplification, scalable profiling method, uneven translation rate, inverse toeprinting
Funders Acknowledgements:
Agence Nationale de la Recherche
Grant ID: ANR-22-CE11- 2 0012-01
Disclaimer
Please note this protocol was originally published on the Protocol Exchange repository; a PDF of the protocol as it appeared there is attached. An automated process was used to import the protocol onto protocols.io. If there are internal references to other steps in the protocol they may not be accurately reflected, please refer to the attached PDF of the protocol to check. Please email us at [email protected] with the DOI of the protocol and any errors that you identify as we can fix these if necessary.
Abstract
Uneven translation rates resulting from the mRNA context, nascent amino acid sequence, or a variety of extraneous factors are key to controlling the expression level, folding, and regulation of the proteome. Although ribosome profiling (Ribo-Seq) is the method of choice to provide genome-wide information on protein synthesis in vivo, tools to systematically study this process in vitro are also needed. To this end, we developed inverse toeprinting coupled to next-generation sequencing (iTP-seq), a scalable profiling method that generates ribosome-protected mRNA fragments without a priori knowledge of the translated sequences (Ref. 1: Seip et al. 2018, Life Science Alliance). Protected mRNA fragments obtained by iTP-Seq consist of the entire coding region upstream of stalled ribosomes, making it possible to work with random or focused transcript libraries rather than sequenced genomes onto which short mRNA footprints must be mapped. We have used iTP-seq to investigate the context dependence of ribosome-targeting antibiotics (Ref. 2: Beckert et al. 2021, Nature Communications; Ref 3: Leroy et al. 2023, Nature Chemical Biology). More generally, iTP-Seq could be used to study any ribosome-related process in which the mRNA sequence or context impact the rate of translation.
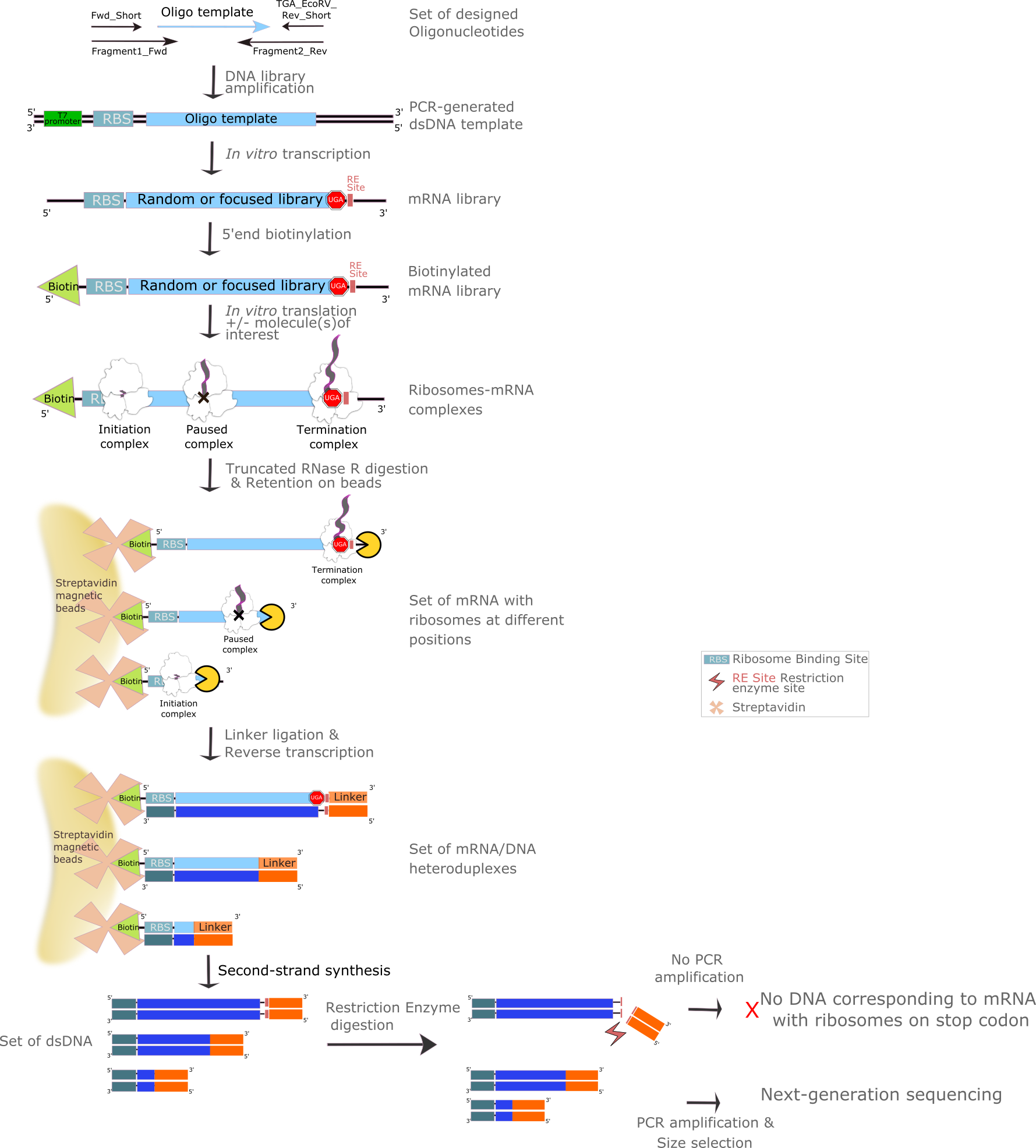
Here, we present a reproducible protocol for iTP-Seq, which substantially reduces the time and effort required to generate sequencing libraries. This protocol consists in 12 main steps: DNA library amplification, in vitro transcription, 5'end mRNA biotinylation, in vitro translation, RNase R digestion, retention on straptavidin-coated beads, linker ligation, reverse transcription, second-strand syntesis, restriction enzyme digestion, PCR amplification and NGS-adapters addition. The time required to complete a single round of iTP-seq is approximately 10 days (Figure 1: Method overview).
Attachments











Manuscript.Pdf
2.1MB
Guidelines
Timing
Steps 1-12, DNA template generation: 4.5 hours
Steps 13-30, In vitro transcription: 5 hours
Steps 31-63, 5'-end biotinylation and assessing biotinylation efficiency: ~ 8 hours
Steps 64-69, In vitro translation: 1 hour
Steps 70-74, RNase R digestion: 45 minutes
Steps 75-91, mRNA purification on Dynabeads M-280: ~2 hours
Steps 92-98, Linker ligation: ~3.5 hours
Steps 99-103, Reverse transcription: 1 hour
Steps 104-121, Fill-Up, restriction enzyme treatment and PCR to amplify cDNA: 7 hours
Steps 122-144, DNA Size Purification of cDNA (gel migration, elution, precipitation): 48 hours
Steps 145-159, Addition of NGS adapters: 8 hours
Steps 160-164, (Optional) Input library preparation for NGS: 5 hours
Steps 165-168 Final library preparation for NGS: 4 hours
Steps 169-, NGS data analysis: 2+ hours
Materials
Reagents:
- Acetic acidMerck MilliporeSigma (Sigma-Aldrich)Catalog #33209-1L Stored at room temperature.
Note
! CAUTION Corrosive.
- Acrylamide/Bis-acrylamide solutionMerck MilliporeSigma (Sigma-Aldrich)Catalog #A9926-5X100ML Stored at 4°C.
Note
! CAUTION Carcinogenic, mutagenic, reprotoxic, irritant and displays acute toxicity. Handle under the fume hood, wear adequate personal protective equipment.
- Ammonium acetateMerck MilliporeSigma (Sigma-Aldrich)Catalog #A1542-500G Stored at room temperature.
Note
! CAUTION Causes skin irritation. Wear adequate personal protective equipment.
- Ammonium chloride ( ≥ 99.5 %)Merck MilliporeSigma (Sigma-Aldrich)Catalog #A9434 Stored at room temperature.
- Ammonium persulfateMerck MilliporeSigma (Sigma-Aldrich)Catalog #A3678-25G Stored at 4°C.
Note
! CAUTION Oxidizing liquid with acute toxicity (oral, dermal, inhalation), skin-eye irritation, inhalation hazard. Handle under the fume hood, wear adequate personal protective equipment.
- ApoI-HF - 1,000 unitsNew England BiolabsCatalog #R3566S Stored at -20°C.
- BCIP/NBT Color Development SubstratePromegaCatalog #S3771 Stored at -20°C.
- 3'-Biotin-GTP - 0.5 umolNew England BiolabsCatalog #N0760S Make aliquots of suitable size to avoid repeated freeze-thawing. Stored at –20°C.
- BIS-TRISMerck MilliporeSigma (Sigma-Aldrich)Catalog #B7535-500G Stored at room temperature.
Note
- ! CAUTION Causes skin irritation. Wear adequate personal protective equipment.
- Bioanalyzer chips and reagents (DNA 1000)Agilent TechnologiesCatalog #5067-1504 Reactives stored at 4°C and chips stored at room temperature.Boric acidMerck MilliporeSigma (Sigma-Aldrich)Catalog #B7901 Stored at room temperature.
Note
- ! CAUTION Reprotoxic. Wear adequate personal protective equipment.
- Bromophenol blueFisher ScientificCatalog #10223280 Stored at room temperature.
- Calcium chlorideEuromedexCatalog #T885 Stored at room temperature.
- Century™-Plus RNA MarkersThermo Fisher ScientificCatalog #AM7145 Stored at -20°C.
- Deoxynucleotide (dNTP) Solution MixNew England BiolabsCatalog #N0447S Stored at -20°C.
- DL-Dithiothreitol solutionMerck MilliporeSigma (Sigma-Aldrich)Catalog #43816-50ML Stored at 4°C
Note
- ! CAUTION Harmful. Causes skin and eye irritation. Use personal protective equipment and handle under fume hood.
- DNA oligonucleotides. Unless indicated otherwise, oligonucleotides were ordered from Eurogentec, SEPOP-desalted or purified with RP-cartridge-Gold‱, resuspended to a final concentration of 100 μM in nuclease-free water and stored at –20°C.
| A | B | |
| Oligonucleotide name | Nucleic acid sequence | |
| Frag1_T7_RBS_ATG_ f | CGATCGAATTCTAATACGACTCACTATAGGGCTTAAGTATAAGGAGGAAAAAATATG | |
| Stop_EcoRV_r | TATATGGATCCTTTTTGATATTGATATCTCATCACACCGAGATCG | |
| Frag1_T7_f | CGATCGAATTCTAATACGACTCACTATAG | |
| EcoRV_r | TATATGGATCCTTTTTGATATTGATA | |
| cDNA_f | GTATAAGGAGGAAAAAATATG | |
| Linker_r | ATT-GAT-GGT-GCC-TAC-AG | |
| Library-specific oligonucleotide (e.g NNN15_random-library) | GGAGGAAAAAATATGNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNGCGATCTCGGTGTGA | |
| Biotin_standard | /5'Biosg/AAAAAAAAAAAAAATTAACTCCATCTAA | |
| 3’_linker_ApoI | /5rAPP/GGTATCTCGGTGTGACTGACTGAAAATTTCTGTAGGCACCATCAAT/ddC | |
| 3’_linker_EcoRV (optional) | /5rAPP/GGTATCTCGGTGTGACTGACTGAGATATCCTGTAGGCACCATCAAT/ddC | |
| NGS_f | AATGATACGGCGACCACCG | |
| NGS_r | CAAGCAGAAGACGGCATACGAG | |
| NGS_adapter_f | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTGTATAAGGAGGAAAAAATATG | |
| NGS_adapter_index_r (the n6 stretch denotes the index sequence) | CAAGCAGAAGACGGCATACGAGATnnnnnnGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCGATTGATGGTGCCTACAG |
- Linkers
Linkers EcoRV and ApoI (Integrated DNA Technologies) are modified as follows:
5’-Adenylated; fully activated and ready for use. The adenylation will enable ligation of the ssDNA to the mRNA.
3’-end blocked with a dideoxy-C base. The ddC will prevent the linker from being circularized.
- Dynabeads M-280 StreptavidinThermo Fisher ScientificCatalog #11205D Stored at 4°C.
- EcoRV-HF - 4,000 unitsNew England BiolabsCatalog #R3195S Stored at -20°C.
- Ethyl alcohol, pureMerck MilliporeSigma (Sigma-Aldrich)Catalog #32221 Stored at room temperature.
Note
- ! CAUTION Flammable. Wear adequate personal protective equipment.
- Ethylenediaminetetraacetic acid disodium salt dihydrateMerck MilliporeSigma (Sigma-Aldrich)Catalog #E5134-500G Stored at room temperature.
Note
! CAUTION Causes skin irritation.
- FormamideMerck MilliporeSigma (Sigma-Aldrich)Catalog #F9037 Stored at room temperature.
Note
- ! CAUTION Reprotoxic and carcinogenic. Wear adequate personal protective equipment.
- L-Glutamic acid monopotassium salt monohydrateMerck MilliporeSigma (Sigma-Aldrich)Catalog #49601-100G Stored at room temperature.
- GlycoBlue™ Coprecipitant (15 mg/mL)Invitrogen - Thermo FisherCatalog # AM9515 Stored at -20°C.
- Hydrochloric acidMerck MilliporeSigma (Sigma-Aldrich)Catalog #30721-M Stored at room temperature.
Note
- ! CAUTION Very corrosive. Wear adequate personal protective equipment and handle under the fume hood.
- Low Molecular Weight DNA Ladder - 100 gel lanesNew England BiolabsCatalog #N3233S Stored at -20°C.
- Magnesium acetate tetrahydrateMerck MilliporeSigma (Sigma-Aldrich)Catalog #M5661-250G Stored at room temperature.
- Magnesium chloride hexahydrateMerck MilliporeSigma (Sigma-Aldrich)Catalog #M2670-1KG Stored at room temperature.
Note
- ! CAUTION Causes skin irritation. Wear adequate personal protective equipment.
- N,N,N′,N′-TetramethylethylenediamineMerck MilliporeSigma (Sigma-Aldrich)Catalog #T9281-25ML Stored at 4°C.
Note
- ! CAUTION Flammable, corrosive, causes skin irritation. Handle under the fume hood, wear adequate personal protective equipment.
- Phusion High-Fidelity DNA Polymerase - 100 unitsNew England BiolabsCatalog #M0530S Stored at -20°C.
- Phusion HF Buffer Pack - 6.0 mlNew England BiolabsCatalog #B0518S Stored at -20°C.
- Potassium hydroxideMerck MilliporeSigma (Sigma-Aldrich)Catalog #P1767-250G Stored at room temperature.
Note
- ! CAUTION Causes skin irritation. Wear adequate personal protective equipment.
- Potassium phosphate monobasic Merck MilliporeSigma (Sigma-Aldrich)Catalog #P9791 Stored at room temperature.
- Potassium phosphate dibasicMerck MilliporeSigma (Sigma-Aldrich)Catalog # 795496 Stored at room temperature.
- Powdered milk (any commercial non-fat dry milk). Stored at room temperature.
- Putrescine dihydrochlorideMerck MilliporeSigma (Sigma-Aldrich)Catalog #P7505 Stored at room temperature.
- Sodium chlorideMerck MilliporeSigma (Sigma-Aldrich)Catalog #S7653-5KG Stored at room temperature.
- Spermidine trihydrochlorideMerck MilliporeSigma (Sigma-Aldrich)Catalog #S2501 Stored at room temperature.
- Streptavidin Alkaline Phosphatase, 0.5mlPromegaCatalog #V5591 Stored at 4°C.
- SuperScript™ III Reverse TranscriptaseThermo Fisher ScientificCatalog #18080044 Stored at -20°C.
- SYBR™ Gold Nucleic Acid Gel Stain (10,000X Concentrate in DMSO)Thermo Fisher ScientificCatalog #S11494 Stored at -20°C.
Note
- ! CAUTION Intercalating agent, mutagenic, carcinogenic. Wear adequate personal protective equipment. Protect from the light.
- T4 RNA Ligase 2, truncated - 10,000 unitsNew England BiolabsCatalog #M0242L Stored at -20°C.
- T7 RiboMAX(TM) Express Large Scale RNA Production SystemPromegaCatalog #P1320 Stored at -20°C.
- Trisodium citrate dihydrateMerck MilliporeSigma (Sigma-Aldrich)Catalog #S1804-500G Stored at room temperature.
- Trizma® baseMerck MilliporeSigma (Sigma-Aldrich)Catalog #T1503-1KG Stored at room temperature.
- RNase RMedChemExpressCatalog #HY-KE7055 Stored at -20°C.
- TWEEN® 20Merck MilliporeSigma (Sigma-Aldrich)Catalog #P1379-100ML Stored at room temperature.
- UreaMerck MilliporeSigma (Sigma-Aldrich)Catalog #U5378-1KG Stored at room temperature.
- WaterMerck MilliporeSigma (Sigma-Aldrich)Catalog #W4502-10X50ML Stored at room temperature.
Note
! CAUTION Causes skin irritation. Handle under the fume hood. Wear adequate personal protective equipment.
- Vaccinia Capping System - 400 unitsNew England BiolabsCatalog #M2080S Stored at -20°C.
- Xylene cyanolBiosolveCatalog #242223 Stored at room temperature.
- All DNA templates are purified using RP-Cartridges, resuspended at 100 mM and stored at -20°C.
- NNS and NNN libraries encode peptides featuring 15 degenerate codons following the initiator methionine (S = C or G; N = any of the four dNTPs). Other custom libraries of varying complexities may be generated for various purposes. Libraries are resuspended at 100 mM and stored at -20°C.
KITS:
- QIAquick Spin ColumnsQiagenCatalog #28115
- RNA Clean & Concentrator™-5Zymo ResearchCatalog #R1015 or R1016)
- PURExpress RF123 Kit - 10 rxnsNew England BiolabsCatalog #E6850S ). Stored at –80°C.
Equipment:
- Bioanalyzer 2100 (Agilent Technologies, Cat. No. G2939A).
- CBS “Lite” Vertical Electrophoresis System (CBS Scientific, Cat. No. SG-250) or system with equivalent plate size (W x L: 16.5 x 22 cm).
- DNA 1000 Kit (Agilent Technologies, Cat. No. 5067-1504).
- EV261 Power Supply (Consort‱, Cat. No. EV261).
- FiveEasy Plus pH meter FP20 (Mettler Toledo‱, Cat. No. 30266627).
- Gel Doc‱ XR+ System (Bio-Rad, Cat. No. 170-8195) or equivalent. A blue light transilluminator can also be used.
- Heraeus Multifuge X3R (Thermo Scientific, Cat. No. 75004515).
- Heratherm Heating and Drying Oven (Thermo Scientific, Cat. No. OGH60-S).
- Amersham‱ Hybond-N+ membrane (20 ×20 cm) (Cytiva, Cat. No. RPN2020B).
- IKA‱ Vortex 3 (IKA, Cat. No.0003340000).
- Mini-PROTEAN‱ Tetra Vertical Electrophoresis Cell (Bio-Rad, Cat. No. 658000EDU) or system with equivalent plate size (W x L: 8.3 x 7.3 cm).
- miVac DNA Concentrator (Fisher Scientific, Cat. No. DNA-23050-B00).
- NanoDrop 2000c spectrophotometer (ThermoScientific, Cat. No. ND-2000C).
- PowerPac Basic Power Supply (Bio-Rad, Cat.No. 164-5050).
- RotoFlex Benchtop Tube Rotator (Cole-Parmer‱,Cat. No. 0439733).
- Sorvall Legend Micro 21R Centrifuge (Thermo Scientific, Cat. No. 75002446).
- Sprout‱ Plus Mini Centrifuge (Heathrow Scientific, Cat. No. 120610).
- T100 Thermal cycler (Bio-Rad, Cat. No.186-1096).
- ThermoMixer‱ C (Eppendorf, Cat.No. 5382000015).
- 16-Tube SureBeads‱ Magnetic Rack (Bio-Rad, Cat. No. 1614916).
- Vivaspin‱ 6 Centrifugal Concentrators 0.2 μm (Sartorius, Cat. No. VS0672).
DNA TEMPLATE GENERATION
Experimental design (Figure 2)
The DNA template contains a T7 promoter (TAATACGACTCACTATAG), a Ribosome Binding Site (RBS - AGGAGG), a sequence of interest (random ORF, etc.), two TGA stop codons, and an EcoRV restriction site located near the stop codons. EcoRI and BamHI restriction sites are present at the 5’ and 3’ ends to allow insertion of the template into a plasmid, if needed. The DNA template is generated by PCR using 5 oligonucleotides: Fwd_short, Fragment1_fwd, Fragment_rev, TGA_EcoRV_rev_short, and a forward oligonucleotide containing the sequence of interest (e.g. GGA-GGA-AAA-AAT-ATG-(NNN)15-GCG-ATC-TCG-GTG-TGA for the NNN-15 library).
TBE gel preparation (TIMING 1.5 hours)
To make 1 or 5 x 15-well (8.3 x 7.3 cm) TBE-acrylamide gel(s) (9% or 12% (wt/vol) depending on the length of the template), mix the following.
| A | B | C | D | E | F | |
| Reagent | Starting concentration | Final concentration | Unit | Volume for 1 gel | Volume for 5 gels | |
| 19:1 Acrylamide:bisacrylamide solution | 40 | 9 or 12 | % wt/vol | 1.35 or 1.8 mL | 6.75 or 9 mL | |
| TBE | 10 | 1 | X | 0.6 mL | 3 mL | |
| Nuclease-free water | 4.05 or 3.6 mL | 20.25 or 18 mL | ||||
| APS | 10 | 0.04 | % wt/vol | 24 μL | 120 μL | |
| Mix by inverting | ||||||
| TEMED | 100 | 0.1 | % vol/vol | 6 μL | 30 μL |
Note
! CAUTION Gels must be prepared under the fume-hood because APS, TEMED and acrylamide are very toxic and irritant.
Mix gently by inverting 5-6 times and pour the gel(s) immediately.
Wait 1 hour for the gel(s) to polymerize. Use gel(s) immediately or wrap in a damp tissue inside an airtight bag. Store at 4°C and use within 2 weeks.
1h
PCR reaction (TIMING 1 hour)
While the gel is polymerizing, prepare samples for the PCR (25 μL per reaction). Mix well. Remember to include suitable PCR controls.
| A | B | C | D | E | |
| Reagent | Starting concentration | Final concentration | Unit | Volume for 1 reaction (μL) | |
| HF Phusion Buffer | 5 | 1 | X | 5 | |
| dNTPs | 10 (each) | 0.2 | mM | 0.5 | |
| Frag1_T7_f primer | 10 | 0.2 | μM | 0.5 | |
| EcoRV_r primer | 10 | 0.2 | μM | 0.5 | |
| Frag1_T7_RBS_ATG_f primer | 1 | 0.02 | μM | 0.5 | |
| Stop_EcoRV_r primer | 1 | 0.02 | μM | 0.5 | |
| Library-specific oligonucleotide | 1 | 0.02 | μM | 0.5 | |
| Phusion Polymerase | 2 | 0.02 | U/μL | 0.25 | |
| Nuclease-free water | 16.75 |
Run the PCR using the following thermocycling parameters.
| A | B | C | |
| Step | Temperature | Time | |
| Initial Denaturation | 98°C | 30 s | |
| 6-24 cycles depends on the template | 98°C | 10 s | |
| 62°C | 5 s | ||
| 72°C | 10 s |
Note
PAUSE POINT Samples can be stored at –20°C until the next step.
Analysis of PCR products by TBE-acrylamide gel electrophoresis (TIMING 1.5 hours)
Mix 1 μL of PCR product, 4 μL of nuclease-free water and 1 μL of 6X Loading Dye.
Load 0.5 μL of Low Molecular Weight DNA Ladder and 5 μL of each sample onto the gel.
Run the gel for 1 hour at 200 V in 1X TBE buffer.
1h
Stain the gel in 1X TBE buffer containing SYBR Gold (1:10000 dilution of stock solution) for 15 minutes.
Note
CRITICAL STEP Do not use the same SYBR Gold solution more than 4 times.
15m
Visualize the stained gel on a transilluminator system (Gel Doc‱ XR+ System or equivalent).
- If the PCR was successful, produce large amounts of DNA for in vitro transcription by scaling up the reaction volume to 1,600 μL (50 μL reactions in 4 strips of 8 tubes). Pool 8 tubes/column for PCR purification.
Note
?TROUBLESHOOTING
| A | B | C | |
| Problem | Possible reason | Possible solution | |
| Low PCR product yields | Low PCR efficiency due to the NNN template | A minimal concentration of 125 ng/mL of template DNA is required for the in vitro transcription step. If the concentration of PCR product is too low, pool multiple PCR reactions into a single QiaQuick PCR purification column and/or use a SpeedVac to concentrate DNA | |
| Byproducts in PCR amplification | Too many PCR cycles can introduce bias and result in larger, non-specific products | We recommend carrying out a pilot experiment first to determine the minimum number of PCR cycles required to amplify the desired targets. The number of cycles should be chosen to obtain the band of interest without any additional products. In cases where the optimum number of PCR cycles is low (e.g. 8 cycles for the NNN15 library), increase the number of reactions accordingly and pool them before purification to obtain adequate DNA yields | |
| PCR amplification in water control | Contaminating DNA is present in the PCR master mix | Repeat the PCR with filter tips and clean reagents, pipettes, and surfaces to minimize contamination |
PAUSE POINT Samples can be stored at –20°C until the next step.
Purify DNA using a QiaQuick PCR Purification Kit or equivalent according to the manufacturer’s instructions. Elute DNA in 30 μL of nuclease-free water.
Note
CRITICAL STEP Wash the column at least twice with PE-buffer and dry the column with an additional centrifugation step.
Determine the concentration of the PCR product using a Nanodrop or equivalent. Pure DNA should have an A260/280 ratio of 1.8 and A260/230 ratio between 1.8 and 2.2.
Note
CRITICAL STEP The DNA template stock solution should have a minimum concentration of 125 ng/μL for the in vitro transcription step. If needed, concentrate the DNA template using a miVac DNA
Concentrator or equivalent.
PAUSE POINT Samples can be stored at –20°C until the next step.
IN VITRO TRANSCRIPTION
Experimental design (Figure 3)
The mRNA is transcribed in vitro from the DNA template yielding an mRNA fragment containing an RBS, a region of interest, spacers, two stop codons (UGA) and the EcoRV restriction site.
TBU Gel preparation (TIMING 1.5 hours)
To make 1 or 5 x 15-well (8.3 x 7.3 cm) TBU-acrylamide gel(s) (9% (wt/vol)), mix
the following.
| A | B | C | D | E | F | |
| Reagent | Starting concentration | Final concentration | Unit | Volume for 1 gel | Volume for 5 gels | |
| Urea | 48 | % wt/vol | 2.88 g | 14.4 g | ||
| 19:1 Acrylamide:bisacrylamide solution | 40 | 9 | % wt/vol | 1.35 mL | 6.75 mL | |
| TBE | 10 | 1 | X | 0.6 mL | 3 mL | |
| Nuclease-free water | Up to 6 mL | Up to 30 mL | ||||
| CRITICAL STEP Mix gently on rotator to completely dissolve the urea | ||||||
| APS | 10 | 0.04 | % wt/vol | 24 μL | 120 μL | |
| Mix by inverting | ||||||
| TEMED | 100 | 0.1 | % vol/vol | 6 μL | 30 μL |
Mix gently by inverting 5-6 times and pour the gel(s) immediately.
Wait 1 hour for the gel(s) to polymerize. Use gel(s) immediately or wrap in a damp tissue inside an airtight bag. Store at 4°C anduse within 2 weeks.
1h
In vitro transcription reaction (TIMING 4 hours)
Note
A small-scale in vitro transcription reaction (20 μL) should be carried out to determine transcription efficiency for every new template, prior to scaling up. As a rule of thumb, 5 pmol of biotinylated mRNA (ideally as a 5 μM stock solution) will then be needed per in vitro translation reaction. This, along with the number of experimental conditions and the number of replicates, should be kept in mind when scaling up the transcription reaction, to ensure that enough RNA can be obtained to perform the entire iTP-Seq experiment.
While the TBU gel is polymerizing, mix the reaction components in a 1.5 mL microtube at room temperature.
| A | B | C | D | E | |
| Reagent | Starting concentration | Final concentration | Unit | Volume for 1 reaction (μL) | |
| RiboMAX Express T7 2X Buffer (contains NTPs) | 2 | 1 | X | 10 | |
| Linear DNA template | - | 1 | μg total | 1-8 | |
| Nuclease-free water | 0-7 | ||||
| Enzyme Mix, T7 Express | - | - | - | 2 |
Note
OBSERVATIONS Frozen RiboMAX‱ Express T7 2X Buffer will contain a precipitate that can be
dissolved by warming the buffer at 37°C and mixing well.
Keep a 1 μL aliquot from the reaction at t=0 and DNA template as controls.
3h
Incubate at 37°C in a thermomixer for 3 hours.
Spin down the reaction to pellet the pyrophosphate resulting from nucleotide incorporation for 30 minutes at full speed at 4°C. Transfer the supernatant containing the mRNA into a new vial.
Note
CRITICAL STEP Do not freeze transcription reactions. After transcription is complete, proceed directly to the next step.
30m
DNase treatment of in vitro transcribed mRNA (TIMING 30 minutes)
Note
CRITICAL STEP All following steps should be carried out under RNase-free conditions.
Add 1 μL of RQ1 RNase-free DNase (1U/μL stock).
Incubate for 15 minutes at 37°C in a thermomixer.
15m
Purification and gel analysis of in vitro transcribed mRNA (TIMING 30 minutes)
Purify mRNA using a RNA Clean & Concentrator‱-5 Kit or equivalent according to the manufacturer’s instructions. If the sample consists of RNA species 17-200 nt only, use 1.5 volumes of ethanol for the
binding step. Elute DNA in 15 μL of nuclease-free water.
Determine the concentration of the mRNA using a Nanodrop or equivalent. Pure mRNA should
have an A260/280 ratio of 2 and A260/230 ratio between 1.8 and 2.2.
Pre-run the 9% (wt/vol) TBU-Acrylamide gel for about 20 minutes at 200 V in 1X TBE.
Mix a volume of mRNA corresponding to 25-50 ng with 15 μL of RNA loading buffer.
Heat samples and Century-Plus RNA Marker for 5 minutes at 95°C.
Load 15 μL of Century-Plus RNA Marker and the entire amount of each sample onto the gel.
Note
CRITICAL STEP Remember to flush out the wells with a pipette or syringe just prior to loading your samples in order to remove urea.
Run the gel for 1 hour at 200 V in 1X TBE buffer.
1h
Stain the gel in 1X TBE buffer containing SYBR Gold (1:10000 dilution of stock solution) for 15 minutes.
15m
Visualize the stained gel on a transilluminator system (Gel Doc‱ XR+ System or equivalent).
Note
CRITICAL STEP Do not use the same SYBR Gold solution more than 4 times.
! CAUTION Clean electrophoresis plates with soap and rinse with distilled water. Never use
ethanol.
? TROUBLESHOOTING
| A | B | C | |
| Problem | Possible reason | Possible solution | |
| Low mRNA yields | Low efficiency of in vitro transcription | Increase the concentration of template DNA in the reaction mixture | |
| Smeary TBU-acrylamide gel | A poorly-resolved TBU-acrylamide gel may result from sample overloading | If a smear is observed instead of a clear band, load a smaller volume of the purified mRNA sample on the gel |
PAUSE POINT Samples can be stored at –20°C until the next step.
5’-BIOTINYLATION OF mRNA
Experimental design (Figure 4)
In order to biotinylate the 5’-end of mRNA, in vitro transcripts are treated with 3’-biotin GTP (mRNA:3’-Biotin-GTP ratio of 1:100 ) and Vaccina Capping Enzyme. The optimal mRNA:3’-Biotin-GTP ratio for biotinylation is 1:100. Usually, 200 pmol of mRNA can be biotinylated with 20,000 pmol of 3’-Biotin-GTP in a 20 μL reaction volume. Keep in mind that 5 pmol of biotinylated mRNA (ideally as a 5 μM stock solution) will be needed per in vitro translation reaction.
5’-capping of mRNA with 3’-Biotin-GTP and Vaccinia Capping Enzyme (TIMING 45 minutes)
In a tube, mix 200 pmol of mRNA template and nuclease-free water up to 12 μL.
Incubate for 5 minutes at 65°C in a thermomixer.
5m
Add the following components to the tube containing the mRNA template.
| A | B | C | D | E | |
| Reagent | Starting concentration | Final concentration | Unit | Volume for 1 reaction (μL) | |
| Capping Buffer | 10 | 1 | X | 2 | |
| 3’-Biotin-GTP | 5 | 1 | mM | 4 | |
| Vaccinia Capping Enzyme | 20 | 2 | U/μL | 2 |
Incubate for 30 minutes at 37°C in a thermomixer.
30m
Purification of biotinylated mRNA (TIMING 30 minutes)
Purify mRNA using an RNA Clean & Concentrator‱ -5 Kit or equivalent according to the manufacturer’s instructions. Elute DNA in 15 μL of nuclease-free water.
Determine the concentration of the mRNA product using a Nanodrop or equivalent. Pure mRNA should have an A260/280 ratio of 2 and A260/230 ratio between 1.8 and 2.2.
Estimation of biotinylation efficiency (TIMING 6 hours) Set the oven to 80°C.
Cut out a piece of Hybond-N+ membrane, enough for 5-6 spots of the standard plus
the number of samples to analyze. Leave a minimum of 0.5 cm between spots.
Note
CRITICAL STEP Do not touch the membrane with your hands. Instead, use tweezers to handle it.
Pre-wet the membrane in 6X SSC buffer (can be reused) for 10 minutes.
10m
In the meantime, prepare dilutions of the biotinylated DNA standard (“Biotin_standard” oligonucleotide) and of the mRNA library.
Note
CRITICAL STEP Dilutions must be done in 6X SSC buffer.
Prepare the following Biotin_standard dilution (Ref1).
| A | B | C | D | E | |
| Starting concentration | Final concentration | Unit | Volume (μL) | ||
| Biotin_standard | 100 | 10 | pmol/μL | 0.5 | |
| 6X SSC Buffer | - | - | - | 4.5 |
Proceed to serial dilutions as follows.
| A | B | C | D | |
| Diluted solution Ref n-1 (μL) | 6X SSC buffer (μL) | Final concentration (pmol/μL) | ||
| Ref2 | 2.5 | 2.5 | 5 | |
| Ref3 | 2.5 | 2.5 | 2.5 | |
| Ref4 | 2.5 | 2.5 | 1.25 | |
| Ref5 | 2.5 | 2.5 | 0.625 |
Prepare the following biotinylated mRNA dilution (Sam1).
| A | B | C | D | E | |
| Starting concentration | Final concentration | Unit | Volume (μL) | ||
| Biotinylated mRNA | - | 5 | pmol/μL | - | |
| 6X SSC Buffer | - | - | - | Up to 5 |
Proceed to serial dilutions as follows.
| A | B | C | D | |
| Diluted solution Sam n-1 (μL) | 6X SSC buffer (μL) | Final concentration (pmol/μL) | ||
| Sam2 | 2.5 | 2.5 | 5 | |
| Sam3 | 2.5 | 2.5 | 2.5 | |
| Sam4 | 2.5 | 2.5 | 1.25 | |
| Sam5 | 2.5 | 2.5 | 0.625 |
Using a pencil, draw a grid on a piece of Whatman paper corresponding to the size of the membrane.
Place the pre-wetted membrane onto the grid.
Using a ballpoint pen to write on the membrane, mark the corners of the grid (visible through the membrane) and an X in one corner.
Pipette the standard and sample dilutions onto the membrane according to the grid.
Note
CRITICAL STEP Do not touch the membrane with the pipette tip and keep the remaining samples for analysis on a TBU gel.
Cover the membrane with a second piece of Whatman paper, wrap in aluminum foil and bake at 80°C for 2 hours.
2h
Place membrane in a 50 mL Falcon tube and block the membrane using TBS-T-Milk. Incubate for 1 hour on a tube rotator at room
temperature.
Note
CRITICAL STEP Ensure that the whole membrane is covered with the solution.
PAUSE POINT Samples can be kept in TBS-T-Milk overnight.
1h
Discard the TBS-T-Milk solution.
Incubate the membrane in 12.5 mL of a 1:2500 dilution of Streptavidin-alkaline Phosphatase in TBS-T buffer for 1 hour on a rotating wheel.
1h
Discard the Streptavidin-alkaline Phosphatase solution.
Wash the membrane for 3 x 10 minutes with TBS-T buffer.
For detection, mix 10 mL of Alkaline Phosphatase buffer and 66 μL of NBT from the NBT/BCIP
detection kit. Mix by inverting and add 33 μL of BCIP. Mix by inverting.
Note
CRITICAL STEP Always mix components in this order, otherwise detection will not work.
Check the labelling after 10–20 minutes.
20m
Discard detection solution and wash membrane once or twice with TBS-T to stop the reaction.
Take a picture of the spots soon after detection.
Note
?TROUBLESHOOTING
| A | B | C | |
| Problem | Possible reason | Possible solution | |
| Low biotinylation efficiency | Low efficiency of vaccinia capping enzyme | Repeat the biotinylation procedure on the same mRNA |
(Optional) Analysis of biotinylated mRNA (TIMING 2 hours)
Pre-run a 9% (wt/vol) TBU-Acrylamide gel for about 20 minutes at 200 V in 1X TBE.
20m
Mix a volume corresponding to 25-50 ng of diluted biotinylated mRNA solution used for the Dot Blot with 15 μL of RNA Loading Buffer.
Heat samples and Century-Plus RNA Marker (15 μL) for 5 minutes at 95°C.
Note
CRITICAL STEP Remember to flush out the wells with your pipette tip or a syringe just prior to loading your samples.
5m
Load 15 μL of Century-Plus RNA Marker and the entire amount of each sample.
Run the gel for 1 hour at 200 V in 1X TBE buffer.
1h
Stain the gel in 1X TBE buffer containing SYBR Gold (1:10000 dilution of stock solution) for 15 minutes.
Note
CRITICAL STEP Do not use the same SYBR Gold solution more than 4 times.
15m
Visualize the stained gel on a transilluminator system (Gel Doc‱ XR+ System or equivalent).
Clean electrophoresis plates with soap and rinse with distilled water.
IN VITRO TRANSLATION
Experimental design (Figure 5)
We use the PURExpress® system to translate biotinylated mRNAs in vitro. This reconstituted protein expression system is based on the PURE system (Ref.4: Shimizu, Y et al. 2001. Nat Biotechnol) and contains all of the components needed for in vitro transcription and translation.
In vitro translation reaction (TIMING 1 hour )
Add 1μL of mRNA (5 μM stock eq. 5 pmol) to each reaction tube.
Prepare an in vitro translation master mix in the order specified by the manufacturer.
Note
OBSERVATIONS There are different types of PURExpress kits. Use the one most suitable for your experiments and refer to its specific protocol. Note that the choice of release factors to use will depend on the nature of the stop codons present in your library. Release factor (RF)1 recognizes UAA and UAG. RF2 recognizes UAA and UGA. RF3 helps RF1 and RF2 to dissociate from the ribosome.
CRITICAL STEP
- Thaw kit solutions on ice just before use and keep on ice at all times. When receiving a new kit, make suitable aliquots of each kit component to avoid repeated freeze-thawing. Freeze unused aliquots in liquid nitrogen and store at –80°C.
- Never spin PURExpress components in a table-top centrifuge. Use a minicentrifuge instead.
- Antibiotics, small-molecule ligands or protein factors may be added to the mix according to your needs. Small molecules are typically first dried inside the tube using a miVac DNA Concentrator. Include control reactions lacking these additional components.
- Unless otherwise specified, volumes, quantities and timings indicated for all subsequent steps correspond to a single replicate of a given condition. Scale accordingly and make pre-mixes where appropriate.
- A 5 μL PURExpress reaction contains 12 pmol of ribosomes. Use 5 pmol of mRNA to ensure an excess of ribosomes over mRNA.
Add 4 μL of in vitro translation reaction mixture to tubes containing mRNA +/– antibiotic, small molecule or any other factor whose effect on translation is to be monitored.
Incubate reactions for 30 minutes at 37 °C in a thermomixer.
Immediately after incubation, place the in vitro translation reactions on ice.
Note
CRITICAL STEP Move on to the next step immediately.
RNASE R DIGESTION
Experimental design (Figure 6)
Truncated RNase R degrades mRNA from the 3’-end. When RNase R encounters the leading ribosome on an mRNA, it drops off the mRNA and a ribosome-protected inverse to eprint is generated.
Note
CRITICAL STEP Adding ice-cold buffer to increase the Mg2+ concentration to 50 mM stabilizes ribosomes on the mRNA. Think to thaw this buffer on ice 1 hour before you use it and make sure it is cold upon addition.
RNase R reaction (TIMING 45 minutes)
Add the following components to the tube containing the translation reaction.
| A | B | C | D | E | |
| Reagent | Starting concentration | Final concentration | Unit | Volume for 1 reaction (μL) | |
| RNase R reaction buffer | 10 | 1 | X | 1.1 | |
| Nuclease-free water | 4.4 | ||||
| RNase R enzyme | 20 | 0.9 | U/μL | 0.5 |
Mix by pipetting and avoid foaming the sample.
Incubate for 30 minutes at 37°C in a thermomixer.
30m
Resuspend digested mRNA in 140 μL of 1X BWT buffer used in the following step. The salt contained in this buffer will ensure that the ribosomes dissociate, and inhibit RNase R. There is no need for further purification.
Note
PAUSE POINT Samples can be stored at –20°C until the next step.
mRNA PURIFICATION ON DYNABEADS M-280
Experimental design (Figure 7)
Streptavidin-coated Dynabeads are used to specifically fish out the biotinylated mRNA.
Note
CRITICAL STEP Always use low binding tubes and tips to prevent Dynabeads from sticking to the tube or pipette tip.
Washingof Dynabeads and mRNA binding TIMING ~2h for up to 12 samples)
Note
OBSERVATIONS The maximal binding capacity of M-280 Dynabeads is ~23 pmol per 10 μL.
1 M NaCl is necessary for minimize non-specific of binding to the beads.
1 wash step consists of :
- Resuspending the beads
- Placing the tube on a magnetic rack
- Waiting for 1 minute
- Removing the supernatant
CRITICAL STEP
- All steps are performed at room temperature and incubation of Dynabeads is always performed on a tube rotator (Rotoflex) to stop the beads from settling at the bottom of the tube.
- Wash tubes one by one to avoid drying the samples and to prevent cross-contamination.
Vortex Dynabeads for at least 30 seconds in order to resuspend them.
Dispense a suitable volume of Dynabeads into a fresh tube (use 5 μL of bead suspension per in vitro translation reaction).
Add an equal volume (or at least 500 μL) of 1X BWT buffer and mix well.
Place the tube on a magnetic rack and wait for at least 1 minute while beads collect on the side of the tube.
Leaving the tube on the magnetic rack, carefully remove the supernatant by pipetting on the side of the tube opposite to where the beads are collected.
Remove the tube from the magnetic rack and resuspend the beads in 500 μL of 1X BWT buffer.
Perform two additional washes with 500 μL of 1X BWT buffer.
Resuspend beads in 1X BWT buffer in order to have 50 μL of bead mixture per translation reaction.
Mix ~150 μL of biotinylated mRNA sample with 50 μL of washed Dynabeads.
Resuspend thoroughly and incubate at room temperature on a tube rotator (Rotoflex) for 15 minutes.
15m
Wash beads twice with 500 μL of 1X BWT buffer.
Wash beads twice with 500 μL of nuclease-free water.
Remove all liquid from the tube after the last wash step.
Perform a quick spin in a minicentrifuge and put on magnetic rack for 1 minute to remove all of the residual liquid.
1m
Resuspend the beads in 4 μL of nuclease-free water. Together with the volume of beads this will give a combined volume of 4.5 μL.
Note
PAUSE POINT Samples can be stored for several days at 4°C. The shorter they stay in the fridge, the better.
LINKER LIGATION
Experimental design (Figure 8)
- The linker is adenylated at its 5’-end and carries ddC at its 3’-end. The adenylation enables ligation of the ssDNA to the mRNA, whereas the ddC stops the linker from being circularized.
- Adding the linker to the 3’-end of the purified mRNA introduces a known 3’-sequence needed for subsequent reverse transcription.
Note
CRITICAL STEP For the linker ligation step, make a pre-mix, ensuring proper mixing of all component in the reaction tube, especially 50% PEG 8000, buffers and DTT.
OBSERVATIONS Be sure to use the correct linker. For example, if there is an EcoRV
restriction site in your construct, use the linker containing the ApoI restriction site (3’_linker_ApoI) and vice versa (3’_linker_EcoRV). Alternating between these two linkers makes it possible to perform successive iTP-Seq cycles.
Ligation (TIMING 2.5 hours)
Prepare the ligation reaction as follows.
| A | B | C | D | E | |
| Reagent | Starting concentration | Final concentration | Unit | Volume for 1 reaction (μL) | |
| 3’_linker_ApoI | 10 | 1 | μM | 1 | |
| T4 RNA ligase 2, truncated Buffer | 10 | 1 | X | 1 | |
| PEG 8,000 | 50 | 15 | % wt/vol | 3 | |
| T4 RNA ligase 2, truncated | 200 | 9 | U/μL | 0.5 |
Dispense 5.5 μL of pre-mix per 1.5 mL microtube containing 4.5 μL of mRNA-on-beads and mix thoroughly.
Incubate the reaction on a tube rotator for 2 hours at room temperature.
Washing (TIMING ~1h for 12 samples)
Wash the beads twice with 500 μL of nuclease-free water.
Remove all liquid from the tube after the last washing step.
Spin tubes in a minicentrifuge and put on a magnetic rack for 1 minute to remove all residual liquid.
1m
Resuspend the beads in 11.5 μL of nuclease-free water.
REVERSE TRANSCRIPTION
Experimental design (Figure 9)
The reverse transcription step generates the DNA complementary to the mRNA (cDNA). It will be possible to perform a PCR on this cDNA to amplify the ribosome-protected sequences.
Reverse Transcription (TIMING 1 hour)
Prepare a pre-annealing pre-mix as follows.
| A | B | C | D | E | |
| Reagent | Starting concentration | Final concentration | Unit | Volume for 1 reaction (μL) | |
| dNTPs | 10 (each) | 0.5 | mM | 1 | |
| Linker_r primer | 2 | 0.1 | μM | 1 |
Dispense 2 μL of the pre-mix to the ~12 μL solution containing the Dynabeads and mix thoroughly.
Incubate for 5 min at 65 °C with 500 rpm shaking in a thermomixer to anneal the primer to its complementary sequence and place the tube(s) on ice.
5m
Prepare a reverse transcription pre-mix as follows.
| A | B | C | D | E | |
| Reagent | Starting concentration | Final concentration | Unit | Volume for 1 reaction (μL) | |
| First Strand synthesis buffer | 5 | 1 | X | 4 | |
| DTT | 100 | 5 | mM | 1 | |
| SuperScript III Reverse Transcriptase | 200 | 10 | U/μL | 1 |
Dispense 6 μL of the pre-mix to each tube and mix thoroughly.
CRITICAL STEPIncubate for 30 minutes at 55°C at 500 rpm in a thermomixer.
Note
CRITICAL STEP To prevent settling of the Dynabeads over time, mix by pipetting after ~15 minutes.
30m
SECOND-STRAND SYNTHESIS, RESTRICTION DIGESTION AND PCR AMPLIFICATION
Experimental design (Figure 10)
cDNA corresponding to inverse toeprints are amplified in 3 steps:
* Second-strand synthesis reaction with the forward primer in order to synthetize the sense strand.
* +/- Restriction Enzyme treatment:
- When ribosomes stall before reaching the stop codon, the restriction site is digested by RNase R and the linker is still present after digestion. Amplification is possible by PCR and the product will be shorter than the full-length mRNA.
- When ribosomes are stalled on the stop codon due to the absence of release factor 2 (RF-2) in the translation reaction, the restriction site is protected from digestion by RNase R. The restriction enzyme can cut the DNA and the linker will be released. Consequently, cDNA will not be amplified.
- If no restriction enzyme is used, cDNAs corresponding to full-length mRNAs will also be amplified.
*PCR:
cDNA that has not undergone restriction digestion is amplified.
To determine the correct number of PCR cycles for amplification, split the 50 μL reaction into 4 x 12.5 μL tubes and test different numbers of PCR cycles (e.g. 8, 10, 12, 14). CRITICAL STEP This step is important to choose the minimum number of PCR cycles required to see your product, but not the higher molecular weight contaminants / byproducts.
Second-strand synthesis for PCR optimization step (TIMING 30 minutes)
Prepare the fill-up reaction mix as follows.
| A | B | C | D | E | |
| Reagent | Starting concentration | Final concentration | Unit | Volume for 1 reaction (μL) | |
| HF Phusion Buffer | 5 | 1 | X | 10 | |
| dNTPs | 10 (each) | 0.2 | mM | 1 | |
| cDNA_f primer | 10 | 0.2 | μM | 1 | |
| Phusion Polymerase | 2 | 0.02 | U/μL | 0.5 | |
| Nuclease-free water | 35.5 |
Dispense 48 μL of the fill-up reaction mix into a PCR tube containing 1 μL of reverse transcription product (Dynabeads mix).
Perform the fill-up reaction using the following thermocycling parameters.
| A | B | C | |
| Step | Temperature | Time | |
| Initial Denaturation | 98°C | 10 s | |
| Annealing | 42°C | 10 s | |
| Elongation | 72°C | 30 s |
PCR optimization (TIMING 1 hour)
Add 1 μL of the Linker_r primer (10 μM stock). Split your PCR reaction mix into 4 x 12.5 μL tubes.
Run the PCR using the following thermocycling parameters.
| A | B | C | |
| Step | Temperature | Time | |
| Initial Denaturation | 98°C | 30 s | |
| 10-18 cycles CRITICAL STEP Number of cycles depends on the sample | 98°C | 10 s | |
| 42°C | 10 s | ||
| 72°C | 10 s |
Analysis of PCR products by TBE-acrylamide gel electrophoresis (TIMING 1.5 hours)
Mix 3-10 μL of each PCR product with nuclease-free water up to a final volume of 10 μL and add 2 μL of 6X Loading Dye.
Load 0.5 μL of Low Molecular Weight DNA Ladder and 12 μL of each sample onto a TBE gel.
Run the gel for 1 hour at 200 V in 1X TBE buffer.
Stain the gel in 1X TBE buffer containing SYBR Gold (1:10000 dilution of stock solution) for 15 minutes.
Note
CRITICAL STEP Do not use the same SYBR Gold solution more than 4 times.
Visualize the stained gel on a transilluminator system (Gel Doc‱ XR+ System or equivalent).
Second-strand synthesis (TIMING 2 hours)
Once the optimal number of PCR cycles is established, prepare samples for the PCR (5
x 25μL per sample).
| A | B | C | D | E | |
| Reagent | Starting concentration | Final concentration | Unit | Volume for 1 reaction (μL) | |
| HF Phusion Buffer | 5 | 1 | X | 5 | |
| dNTPs | 10 (each) | 0.2 | mM | 0.5 | |
| cDNA_f primer | 10 | 0.2 | μM | 0.5 | |
| Phusion Polymerase | 2 | 0.02 | U/μL | 0.25 | |
| Nuclease-free water | 17.25 |
Dispense 23.5 μL of the fill-up reaction mix into a PCR tube containing 0.5 μL of reverse transcription product (Dynabeads mix).
Run the fill-up reaction using the following thermocycling parameters.
| A | B | C | |
| Step | Temperature | Time | |
| Initial Denaturation | 98°C | 10 s | |
| Annealing | 42°C | 10 s | |
| Elongation | 72°C | 30 s |
Restriction digestion (TIMING 1 hour)
Pool the 5 tubes for each sample.
Add 2.5 μL of restriction enzyme to the PCR mix. If you used a linker with an ApoI site in step 86, use EcoRV-HF in this step and vice-versa.
Mix thoroughly by pipetting.
1h
Incubate for 1 hour at 37 °C in a thermocycler
Add 2.5 μL of the Linker_r primer (10 μM stock) and split into 5 x 25 μL tubes.
Run the PCR using the following thermocycling parameters.
| A | B | C | |
| Step | Temperature | Time | |
| Initial Denaturation | 98°C | 30 s | |
| Number of cycles determined in steps 112-120 | 98°C | 10 s | |
| 42°C | 10 s | ||
| 72°C | 10 s |
DNA SIZE SELECTION AND RECOVERY
18h
Preparation of medium-sized TBE-acrylamide gel and gel electrophoresis (TIMING 20 hours)
Clean two 16.5 x 22 cm glass plates thoroughly with nuclease-free water, ethanol and nuclease-free water and dry them with Kimwipes‱.
Assemble the blue silicone rubber joint, 1.5 mm spacers and the two plates according to the manufacturer’s instructions.
To make a 60 mL medium-sized 9% (wt/vol) TBE-acrylamide gel, mix the following.
| A | B | C | D | E | |
| Reagent | Starting concentration | Final concentration | Unit | Volume for a 60 mL-gel | |
| 19:1 Acrylamide:bisacrylamide solution | 40 | 9 | % wt/vol | 13.5 mL | |
| TBE | 10 | 1 | X | 6 mL | |
| Nuclease-free water | 40.5 mL | ||||
| APS | 10 | 0.04 | % wt/vol | 240 μL | |
| Mix by inverting | |||||
| TEMED | 100 | 0.1 | % vol/vol | 60 μL |
Mix gently by inverting 5-6 times, immediately pour the gel, insert a 20-well comb and let it polymerize.
Mix 25 μL of each sample with 5 μL of 6X loading dye, and load onto the gel. Put side-by-side gels with and without antibiotics or ligands to further identify the band of interest. Load 2 μL of Low Molecular Weight DNA Ladder. Fill empty wells with 5 μL of 6X loading dye.
Note
CRITICAL STEP To minimize cross-contamination, leave empty wells between each type of sample and between the samples and the ladder.
18h
Check that the gel tank is level with the ground. Separate the PCR products on the medium-sized TBE-acrylamide gel by running it at 175V for 18 hours.
Recovering the bands of interest (TIMING 1 hour for 1 gel)
Separate the gel plates and cut a corner to help orient the gel.
Stain the gel in 1X TBE buffer containing SYBR Gold (1:10000 dilution of stock solution) for 15 minutes.
Note
! CAUTION To minimize cross-contamination, rinse the tank with distilled water and ethanol, use fresh SYBR Gold solution and put Saran wrap on the transilluminator plate. Prepare 1 fresh scalpel, 1 syringe and 1 x 15 mL Falcon tube per sample.
Visualize the stained gel on a transilluminator system (Gel Doc‱ XR+ System or equivalent). Take a picture in order to see which part of the gel you will be cutting.
Note
! CAUTION Use a Plexiglas screen for UV protection if you're using a UV transilluminator.
On the transilluminator, cut out the band(s) of interest with a clean scalpel.
Note
CRITICAL STEP If you are using an NNN or NNS library, cut the gel to include the region between the top edge of the start codon band and the stop codon band. Including too much of the start codon will yield too many reads for initiation complexes at the expense of reads for elongation complexes.
Remove the plunger from a 5 mL syringe, put the gel pieces into the syringe, and put the plunger back in.
Take a picture of the gel after cutting the bands out to ensure that you took all of the DNA of interest.
Crush the gel pieces by pushing them through the opening of the syringe and store them in a 15 mL Falcon tube.
Add 10 mL of gel extraction buffer in Falcon tubes containing gels debris. Rinse the syringe, plunger and scalpel with this buffer to avoid losing part of your samples.
Extract the DNA by diffusion (TIMING 24 hours)
Incubate gel fragments at room temperature on a tube rotator (Rotoflex) for at least 24 hours.
Pre-wet a 0.22 μm Vivaspin filter with 6 mL of gel extraction buffer and centrifuge for 1 minute at 4,000 x g at 4°C.
1m
Pour half the solution (~5 mL) containing the gel fragments into a 0.22 μm Vivaspin filter and spin for 5 minutes at 4,000 x g. Keep the flow-through and pour the remainder of the solution (~5 mL) into the 0.22 μm Vivaspin filter. Pipette any of the liquid left around the gel debris into the column. Spin for 5 minutes at 4,000 x g and pool the two flow-throughs.
Note
! CAUTION Tubes and samples are contaminated with SYBR Gold. Use good laboratory practice for handling and disposal of samples.
PAUSE POINT Samples can be stored at –20°C until the next step.
Isopropanol precipitation of inverse-toeprints (TIMING 30 minutes)
Split each ~10 mL sample into 2 mL low-binding Eppendorf tubes (~1.5 mL per tube).
Concentrate DNA using a miVac DNA Concentrator (water mode at 50°C) to reach a volume of ~200 μL in each tube (expected time of evaporation: ~3 hours).
Pool fractions of all samples together (~1.1 mL) and add GlycoBlue‱ Coprecipitant at a final concentration of 50 μg/mL.
Add 1 volume of isopropanol.
Vortex and incubate on ice for 15 minutes.
15m
Store at –80°C overnight.
Recovery of precipitated DNA (TIMING 2 hours)
Pellet DNA by centrifuging at 20,000 x g for 30 minutes at 4°C.
30m
Dry the pellet for ~1 hour on the bench after carefully discarding the supernatant and dry tube walls with clean Kimwipes‱.
1h
Resuspend the DNA pellet in 20 μL of nuclease-free water.
Note
PAUSE POINT Samples can be stored at –20°C until the next step.
ADDITION OF NGS ADAPTERS
Experimental design (Figure 11)
- The goal of this step is to add TruSeq DNA adapters by PCR. The forward primer is universal while the reverse primer contains a specific barcode. DNA from each experimental condition will be amplified with a specific barcoded reverse oligonucleotide, enabling multiplexing during next-generation sequencing (Illumina).
- All samples will be mixed in the desired molar ratios prior to next-generation sequencing.
Note
CRITICAL STEP
- PCR amplification of cDNA should be optimized by running different numbers of cycles (e.g. 12, 14, 16, 18). Previous experiments with NNN or NNS libraries showed that running fewer PCR cycles yields more specific PCR products with the expected size distribution.
- Carefully choose the barcoded reverse primers such that 1 sample is assigned specific barcode.
- Carefully follow Illumina recommendations for barcode design and ensure that there are at least 2 nucleotide differences between the indices to minimize index misassignment.
PCR optimization (TIMING 1.5 hours)
Prepare samples for the PCR (50 μL per reaction). Mix well. Remember to include suitable PCR controls.
| A | B | C | D | E | |
| Reagent | Starting concentration | Final concentration | Unit | Volume for 1 reaction (μL) | |
| HF Phusion Buffer | 5 | 1 | X | 10 | |
| dNTPs | 10 (each) | 0.2 | mM | 1 | |
| NGS_f primer | 10 | 0.2 | μM | 1 | |
| NGS_r primer | 10 | 0.2 | μM | 1 | |
| NGS_ adapter_f primer | 1 | 0.02 | μM | 1 | |
| NGS_adapter_index_r primer | 1 | 0.02 | μM | 1 | |
| Purified cDNA | - | - | - | 0.5 | |
| Phusion Polymerase | 2 | 0.02 | U/μL | 0.5 | |
| Nuclease-free water | 34 |
Split your PCR reaction mix into 4 tubes of 12.5 μL.
Run the PCR using the following thermocycling parameters.
| A | B | C | |
| Step | Temperature | Time | |
| Initial Denaturation | 98°C | 30 s | |
| 10-18 cycles | 98°C | 10 s | |
| 42°C | 10 s | ||
| 72°C | 10 s |
Note
PAUSE POINT Samples can be stored at –20°C until the next step.
Analysis of PCR products by TBE-acrylamide gel electrophoresis (TIMING 2.5 hours)
Prepare a 12% (wt/vol) TBE-acrylamide gel.
Mix 10 μL of PCR reaction with 2 μL of 6X loading dye. Load 0.5 μL of Molecular Weight DNA Ladder and 12 μL of each sample onto the gel.
Run the gel for 1 hour at 200 V in 1X TBE buffer.
1h
Stain the gel in 1X TBE buffer containing SYBR Gold (1:10000 dilution of stock solution) for 15 minutes.
Note
CRITICAL STEP Do not use the same SYBR Gold solution more than 4 times.
15m
Visualize the stained gel on a transilluminator system (Gel Doc‱ XR+ System or equivalent).
Note
! CRITICAL STEP Choose the minimum number of cycles required to see your product, but not the higher molecular weight contaminants / byproducts.
Note
TROUBLESHOOTING
| A | B | C | |
| Problem | Possible reason | Possible solution | |
| Byproducts in PCR amplification | Too many PCR cycles can introduce bias and result in larger, non-specific products | We recommend carrying out a pilot experiment first to determine the minimum number of PCR cycles required to amplify the desired targets. The number of cycles should be chosen to obtain the band of interest without any additional products. In cases where the optimum number of PCR cycles is low (e.g. 8 cycles for the NNN15 library), increase the number of reactions accordingly and pool them before purification to obtain adequate DNA yields | |
| PCR amplification in water control | Contaminating DNA is present in the PCR master mix | Repeat the PCR with filter tips and clean reagents, pipettes, and surfaces to minimize contamination |
Addition of NGS adapters by PCR (TIMING 1.5 hours)
Amplify cDNA within each sample in a 50 μL PCR using the number of cycles determined in step 152.
Note
PAUSE POINT Samples can be stored at –20°C until the next step.
Purification and analysis of PCR products by TBE-acrylamide gel electrophoresis (TIMING 2.5 hours)
Purify DNA using a QiaQuick PCR Purification Kit or equivalent according to the manufacturer’s instructions. Elute DNA in 30 μL of nuclease-free water.
Note
CRITICAL STEP Wash the column at least twice with PE-buffer and dry the column with an additional centrifugation step.
Determine the concentration of PCR product using a Nanodrop or equivalent. Pure DNA should have an A260/280 ratio of 1.8 and A260/230 ratio between 1.8 and 2.2.
Prepare a 12% (wt/vol) TBE-acrylamide gel. Mix a volume of PCR product corresponding to 50 ng, nuclease-free water up to 5 μL and add 1 μL of 6X Loading Dye. Load 0.5 μL of Low Molecular Weight DNA Ladder and 5 μL of each sample onto the gel.
Run the gel for 1 hour at 200 V in 1X TBE buffer.
1h
Stain the gel in 1X TBE buffer containing SYBR Gold (1:10000 dilution of stock solution) for 15 minutes.
Note
CRITICAL STEP Do not use the same SYBR Gold solution more than 4 times.
15m
Visualize the stained gel on a transilluminator system (Gel Doc‱ XR+ System or equivalent).
Note
TROUBLESHOOTING
| A | B | C | |
| Problem | Possible reason | Possible solution | |
| Byproducts in PCR amplification | Too many PCR cycles can introduce bias and result in larger, non-specific products | We recommend carrying out a pilot experiment first to determine the minimum number of PCR cycles required to amplify the desired targets . The number of cycles should be chosen to obtain the band of interest without any additional products. In cases where the optimum number of PCR cycles is low (e.g. 8 cycles for the NNN15 library), increase the number of reactions accordingly and pool them before purification to obtain adequate DNA yields | |
| PCR amplification in water control | Contaminating DNA is present in the PCR master mix | Repeat the PCR with filter tips and clean reagents, pipettes, and surfaces to minimize contamination |
Note
PAUSE POINT Samples can be stored at –20°C until the next step.
(OPTIONAL) INPUT LIBRARY PREPARATION FOR NGS
Note
OBSERVATIONS Sequencing the input mRNA library may be useful to assess whether it has the desired complexity. Although adding NGS adapters directly to the input library is an option, we recommend that you treat the input library in the same manner as other iTP-Seq samples, with the exception of the in vitro translation and RNase R digestions steps. This has the added advantage of taking into account transcription and RT-PCR biases, allowing a direct comparison to be made with the other iTP-Seq samples. To prepare such a library, follow the steps below.
Dilute the biotinylated mRNA library to obtain a stock solution with a concentration of 5 pmol/μL.
Prepare the following reaction mix.
| A | B | C | D | E | |
| Reagent | Starting concentration | Final concentration | Unit | Volume for 1 reaction (μL) | |
| Biotinylated mRNA | 5 | 0.25 | pmol/μL | 1 | |
| In vitro translation buffer | - | - | - | 4 | |
| RNase R reaction buffer | - | - | - | 1.1 | |
| Nuclease-free water | 4.9 | ||||
| BWT buffer | - | - | - | 140 |
Follow the procedure from step 77 to step 114 with subsequent PCR according to step 103 and PCR product purification (steps 11-12) of the input library from mRNA.
5m
Add NGS adapters and proceed to purification and analysis of PCR products following the procedure from step 147 to 161 and 167-168.
PREPARATION OF NGS SAMPLE MIX
1h
Measuring the concentration of PCR products with the Bioanalyzer (TIMING 3 hours)
Assess the quality and quantity of individual DNA libraries for each sample/condition using a Bioanalyzer and a DNA 1000 chip. Please refer to the manufacturer’s recommendations and protocol.
Note
TROUBLESHOOTING
| A | B | C | |
| Problem | Possible reason | Possible solution | |
| Low quality/quantity of NGS samples upon Bioanalyzer analysis | Some indexed NGS adapters could result in less efficient PCR | Use a different primer and/or increase the number of PCR cycles. For samples with low DNA concentration, use High sensitivity DNA chips (Agilent). Since Bioanalyzer chips can occasionally suffer from artefacts, perform 3 independent measurements and use the average value to determine the molar concentration of the DNA library |
Prepare a multiplexed library for NGS. Based on the DNA concentrations determined in the previous step, mix the individual library components according to the desired molar ratios.
Note
CRITICAL STEP The pooling strategy should be optimized based on the specific Illumina platform used and the desired sequencing depth.
Sequencing considerations. The number of replicates and the number of next-generation sequencing reads per sample are also important parameters to consider during the design of an iTP-Seq experiment. An advantage of iTP-Seq is that it requires fewer reads than Ribo-Seq, and a careful design of the sequencing plan can optimize read distribution further to extract maximum information from a deep sequencing run. Each condition should ideally include at least two biological replicates, but we recommend using three or four as this will strengthen statistical confidence in the results. Although we performed our analyses with paired-end sequencing, analysis of a paired-end dataset using only forward reads produced comparable results, suggesting that single-read sequencing may suffice for many applications. To increase the yield of meaningful sequencing reads, we advise discarding the lower portion of the gel band for the translation initiation peak, as inverse toeprints corresponding to initiating ribosomes tend to be overrepresented. For each replicate, 2–3 million reads typically yields optimal results, with approximately half of these falling within the correct range to be analyzed as valid inverse toeprints (i.e. codons 1–10 for an NNN15 library, for which 3-nucleotide periodicity is readily apparent). A greater number of reads decreases the risk of artifacts and false positives; we observed that while experimentally validated features remain consistent, certain sequence combinations appear with lower read counts (~100,000 inverse toeprints), but disappear when analyzed with more than a million inverse toeprints. Finally, it is important to note that the shared sequence at the beginning of all input templates can cause technical issues during sequencing, including reduced cluster diversity and potential base-calling problems. Therefore, we recommend either spiking the sequencing run with PhiX or adding heterogeneity spacers to the index sequence to ensure optimal sequencing quality and cluster identification.
Determining the final library concentration with the Bioanalyzer prior to NGS (TIMING 1 hour)
Assess the quality and quantity of the final library using a Bioanalyzer and a DNA 1000 chip. Place 1 μL of the final NGS library into several wells of the chip to obtain accurate concentrations for NGS. Please refer to the manufacturer’s recommendations and protocol. Fig. 2h shows a representative bioanalyzer quality control analysis.
Wrap parafilm around the cap to prevent sample loss, and send your sample for next-generation sequencing.
NGS DATA ANALYSIS (TIMING 2+ hours)
Retrieve NGS result files from the sequencing facility. These typically consist of files in FASTQ format (two files per sample in case of paired-end reads) and might be compressed (fastq.gz). We usually name the samples with short identifiers. For example, in the case of a study involving an antibiotic such as TcmX we would name noa1 the first untreated replicate and tcx3 the third replicate of a TcmX-treated condition. If relevant we also add the library type (e.g. nnn15) or antibiotic concentration to the sample and file name. A dataset with a control and an antibiotic-treated condition, with 3 replicates each and paired-end sequencing might look like the following:
nnn15_noa1_1.fastq
nnn15_noa1_2.fastq
nnn15_noa2_1.fastq
nnn15_noa2_2.fastq
nnn15_noa3_1.fastq
nnn15_noa3_2.fastq
nnn15_tcx1_1.fastq
nnn15_tcx1_2.fastq
nnn15_tcx2_1.fastq
nnn15_tcx2_2.fastq
nnn15_tcx3_1.fastq
nnn15_tcx3_2.fastq
Process the assembled files to extract the inverse toeprint sequences. In order to do so, install the itpseq package using the following command: pip install itpseq
This should make the itpseq tool available as a command on your machine, together with a Python module for iTP-Seq data analysis.
In the case of paired-end sequencing, pair the reads contained in the two files for each sample (usually the same file prefix with _1 and _2 suffixes) using a dedicated tool. For example, with PEAR35, each pair of files for the nnn15_noa1 sample can be combined into a single assembled file with the following command:
pear -u0 -c126 -f nnn15_noa1_1.fastq -r nnn15_noa1_2.fastq -o nnn15_noa1
This will create a nnn15_noa1.assembled.fastq file, along with additional files for the rejected reads and processing log. The -u0 option sets the maximal proportion of uncalled bases in a read to 0 and -c126 sets the upper bound for the quality score to 126.
Process all of the assembled read files generated in step 165 with the following
command: itpseq parse *.assembled.fastq
By default, the script will extract the sequences between the GTATAAGGAGGAAAAAAT and GGTATCTCGGTGTGACTG adaptor sequences, and retain reads that span 1 to 10 codons from the start codon to the A-site, with a minimum quality of 30. This range does not include the untranslated overhang downstream of the A-site (typically 12–14 nucleotides in E. coli). The default window of 1–10 codons is used because triplet periodicity is generally maintained in this region; however, this parameter can be adjusted based on the experimental data, after visualizing the distribution of inverse-toeprint lengths and inspecting the periodicity. User-defined parameters, including adaptor sequences, codon range, quality cutoff, and untranslated overhang, can be specified as follows:
itpseq parse --left-adaptor GTATAAGGAGGAAAAAAT --right-adaptor GGTATCTCGGTGTGACTG --peptide-size 1-10 --quality 30 --untranslated-overhang 12 *.assembled.fastq
The script will create various files for each input file, which will be used in the subsequent analysis. The nucleotide sequences of the inverse toeprints are exported in [name].nuc.itp.txt, and the matching amino-acid sequences (up to the A-site) in [name].aa.itp.txt.
Generate a PDF report with a default set of graphs and analyses by typing the following command from the directory containing the dataset ("." represents the current directory):
itpseq report .
More advanced exploratory analyses can be performed using the API of the itpseq module using Python scripts and a Jupyter notebook. Examples and tutorials are available in the documentation of the itpseq package and will be periodically updated as novel functionalities are added.
Note
TROUBLESHOOTING
| A | B | C | |
| Problem | Possible reason | Possible solution | |
| Issues with the itpseq package | Documentation does not resolve your issue | Submit a bug report or help request on the GitHub tracker |
Figures
Figure 1. Figure 1. iTP-Seq method overview
Figure 2. Figure 2. DNA template library generation by PCR
Figure 3. Figure 3. mRNA library generation by in vitrotranscription
Figure 4. Figure 4. mRNA library biotinylation by capping 5'end mRNA with Vaccinia Capping Enzyme and biotin-GTP in order to further fish-out mRNA of interest
Figure 5. Figure 5. In vitro translation of biotinylated mRNA library
Figure 6. Figure 6. Digestion from 3'-end to 5'-end of biotinylated mRNA library with RNAse R enzyme
Figure 7. Figure 7. Retention of RNase-R digested mRNA library on Streptavidin-coated Dynabeads
Figure 8. Figure 8. Linker addition on 3'end of mRNA to initiate the reverse transcription
Figure 9. Figure 9. Library cDNA generation by reverse transcription
Figure 10. Figure 10. Double strand DNA generation, EcoRV treatment for removing templates corresponding to ribosome stalled on stop codons and amplification by PCR
Figure 11. Figure 11. Next generation sequencing primers addition to generate the premade library
Protocol references
1. Seip, B. et al. Ribosomal stalling landscapes revealed by high-throughput inverse toeprinting of mRNA libraries. Life Science Alliance. vol. 1 n° 5 (2018).
2. Beckert, B. et al. Structural and mechanistic basis for translation inhibition by macrolide and ketolide antibiotics. Nature Communications. vol.12 n° 4466 (2021).
3.Leroy, EC. et al. Tetracenomycin X sequesters peptidyl-tRNA during translation of QK motifs. Nature Chemical Biology. 19, 1091–1096 (2023).
4. Shimizu, Y., Inoue, A., Tomari, Y. et al. Cell-free translation reconstituted with purified components. Nature Biotechnology. 9, 751–755 (2001).
Associated Publications:
by Britta Seip, Guénaël Sacheau, Denis Dupuy, C Axel Innis
published in Life Science Alliance on 09 Oct, 2018
https://dx.doi.org/10.26508/lsa.201800148