Mar 03, 2022
Immunofluorescence-based assay to assess LRRK2 association with microtubules in HEK293 cells
- Elena Purlyte1,
- Alexia Kalogeropulou1,
- Francesca Tonelli1,
- Dario R Alessi1
- 1Medical Research Council Protein Phosphorylation and Ubiquitylation Unit, School of Life Sciences, University of Dundee, Dow Street, Dundee DD1 5EH, UK



External link: https://doi.org/10.7554/eLife.79771
Protocol Citation: Elena Purlyte, Alexia Kalogeropulou, Francesca Tonelli, Dario R Alessi 2022. Immunofluorescence-based assay to assess LRRK2 association with microtubules in HEK293 cells. protocols.io https://dx.doi.org/10.17504/protocols.io.b5jhq4j6
Manuscript citation:
Vides EG, Adhikari A, Chiang CY, Lis P, Purlyte E, Limouse C, Shumate JL, Spínola-Lasso E, Dhekne HS, Alessi DR, Pfeffer SR, A feed-forward pathway drives LRRK2 kinase membrane recruitment and activation. eLife doi: 10.7554/eLife.79771
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it’s working
Created: February 24, 2022
Last Modified: May 31, 2024
Protocol Integer ID: 58697
Keywords: immunofluorescence microscopy, Laser confocal imaging, LRRK2, microtubules, ASAPCRN, lrrk2 association with microtubule, lrrk2 microtubule, pathogenic lrrk2 mutant, lrrk2 kinase activity, pharmacological inhibition of lrrk2 kinase activity, lrrk2 mutation, relocalization of lrrk2, measuring lrrk2 association, microtubule, lrrk2 association, hek293 cells previous study, confocal immunofluorescence microscopy method, immunofluorescence, hek293 cell, using confocal fluorescence microscopy, confocal fluorescence microscopy, lrrk2, confocal immunofluorescence, fluorescence, electron microscopy
Abstract
Previous studies using confocal fluorescence microscopy and cryo-electron microscopy reported that most pathogenic LRRK2 mutants, as well as pharmacological inhibition of LRRK2 kinase activity with Type-I inhibitors, cause relocalization of LRRK2 to filamentous structures that colocalize with microtubules (PMID: 22080837; PMID: 28453723; PMID: 32783917; PMID: 32814344). Here we describe our confocal immunofluorescence microscopy method for measuring LRRK2 association with microtubules in a cell-based assay. This method can be used to screen the impact that LRRK2 mutations have on LRRK2 microtubule binding, as well as the effect of any compound on LRRK2 association with microtubules.
Attachments

MT assay_zoom_protoc...
23.7MB
Guidelines
This protocol includes our method for:
1) Reverse transfection of HEK293 cells with LRRK2 cDNA;
2) Sample preparation for immunofluorescence microscopy;
3) Imaging and cell counting.
Note:
In parallel with the preparation of samples for immunofluorescence microscopy, we recommend preparing samples for quantitative immunoblotting analysis (as described in dx.doi.org/10.17504/protocols.io.bsgrnbv6) to assess LRRK2 expression levels and efficient LRRK2 inhibition in samples treated with MLi-2.
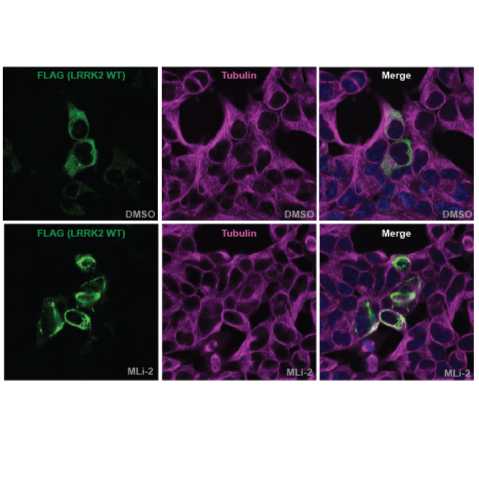
Figure 1: Type I LRRK2 inhibitor MLi-2 enhances LRRK2 filament formation. FLAG-LRRK2 wildtype was transiently expressed in HEK293 cells for 24 hours (for immunoblotting analysis) or 48 hours (for immunofluorescence microscopy analysis). 3 hours prior to lysis or fixation, cells were treated with 100 nM MLi-2 or 0.1% (v/v) DMSO (control vehicle). For immunofluorescence analysis (left panel), cells were fixed with 4% PFA and stained with anti-FLAG (raised in mouse) and anti-tubulin (raised in rabbit) primary antibodies, followed by incubation with anti-mouse Alexa Fluor 488 and anti-rabbit Alexa Fluor 594 secondary antibodies. For immunoblotting analysis cells were harvested in complete lysis buffer and samples subjected to SDS-PAGE and western blotting. Membranes were probed with the indicated antibodies and were developed using the LI-COR Odyssey CLx Western Blot imaging system (right panel).
Materials
Materials and reagents:
- HEK293 cells (ATCC #CRL-1573) cultured in complete growth medium: DMEM (Thermofisher Scientific #11960-044) supplemented with 10% Fetal Calf Serum, qualified, Brazil (Thermofisher Scientific #10270-106), penicillin/streptomycin (Thermofisher Scientific #15140-122) and L-glutamine (Thermofisher Scientific #25030-024).
- N-terminus Flag-tagged LRRK2 wild-type or mutant cDNA and Flag-empty vector cDNA (negative control) in a pCMV5 vector. All plasmids used for our studies are available from the MRC PPU Reagents and Services (https://mrcppureagents.dundee.ac.uk). These include Flag-tagged wild-type LRRK2 (DU6841), G2019S LRRK2 (DU10129), Y1699C LRRK2 (DU26486), I2020T LRRK2 (DU13081) and G2385R LRRK2 (DU27381).
- Polyethylenimine (PEI) “Max” (Linear, Mw 40,000) (Polysciences, Inc., #24765); 1 mg/ml (w/v) solution in milliQ water, pH 7.4; sterile filtered.
- Opti-MEM Reduced Serum Medium (ThermoFisher Scientific #31985062).
- Ibidi µ-Plates (24 Well) - Black ID 14 mm, ibiTreat: #1.5 polymer coverslip, tissue culture treated, sterilized (Ibidi #82426).
- Tissue culture-treated flat bottom cell culture 24-well plates (Thermo Scientific Nunc #142475).
- 4% paraformaldehyde (PFA, Sigma Aldrich #P6148) in PBS, pH 7.4 (make fresh or keep frozen at -20oC until use)
- Phosphate buffered saline (PBS), pH 7.4 (ThermoFisher Scientific #10728775)
- NP-40 alternative (Merck #492016)
- Bovine Serum Albumin (BSA) (Sigma-Aldrich #A7906).
- Primary antibodies: Monoclonal anti-Flag M2 antibody (Sigma Aldrich #F1804); anti-β-tubulin antibody (Abcam ab6046)
- DAPI (bisBenzimide H 33342 trihydrochloride) (Sigma Aldrich #B2261)
- Secondary antibodies: goat anti-mouse Alexa Fluor 488 (ThermoFisher Scientific #A-11029) and goat anti-rabbit Alexa Fluor 594 (ThermoFisher Scientific #A-11012)
- MLI-2 LRRK2 inhibitor (100 micromolar (µM) stock in DMSO) (Available from (https://mrcppureagents.dundee.ac.uk)
- DMSO
- Lysis buffer:
| A | B | |
| Tris-base pH 7.4 | 50 mM | |
| EGTA | 1 mM | |
| 2-Glycerophosphate | 10 mM | |
| Sodium fluoride | 50 mM | |
| Sodium pyrophosphate | 5 mM | |
| Sucrose | 270 mM | |
| Microcystin-LR (Enzo Life Sciences, #ALX-350-012) | 1 mg/ml | |
| Sodium orthovanadate | 1 mM | |
| Complete EDTA-free protease inhibitor cocktail (Roche #11836170001) | ||
| Triton X-100 just before lysis | 1% (v/v) |
Human Embryonic Kidney (HEK293) CellsATCCCatalog #CRL-1573
DMEM high glucose no glutamineThermo Fisher ScientificCatalog #11960044
Gibco™ Fetal Bovine Serum qualified BrazilThermo Fisher ScientificCatalog #10270106
Penicillin/StreptomycinThermo Fisher ScientificCatalog #Invitrogen 15140-122
L-Glutamine (200 mM)Thermo FisherCatalog #25030024
PEI MAX® - Transfection Grade Linear Polyethylenimine Hydrochloride (MW 40000)Polysciences, Inc.Catalog #24765-1
Opti-MEM™ Reduced Serum MediumThermo Fisher ScientificCatalog #31985062
µ-Plate 24 Well Black ID 14 mmIbidiCatalog #82426
Nunc™ Cell-Culture Treated Multidishes, 24 wellThermo FisherCatalog #142475
ParaformaldehydeMerck MilliporeSigma (Sigma-Aldrich)Catalog #P6148
NP-40 AlternativeMerck Millipore (EMD Millipore)Catalog #492016
Bovine Serum Albumin (BSA) Merck MilliporeSigma (Sigma-Aldrich)Catalog #A7906
Monoclonal M2 antibody (anti-FLAG)Merck MilliporeSigma (Sigma-Aldrich)Catalog #F1804-200UG
Anti-beta Tubulin antibody - Loading ControlAbcamCatalog #ab6046
bisBenzimide H 33342 trihydrochlorideMerck MilliporeSigma (Sigma-Aldrich)Catalog #B2261
Goat anti-Mouse IgG (H L) Highly Cross-Adsorbed Secondary Antibody Alexa Fluor 488Thermo Fisher ScientificCatalog #A-11029
Goat anti-Rabbit IgG (H L) Cross-Adsorbed Secondary Antibody Alexa Fluor 594Thermo Fisher ScientificCatalog # A-11012
Microcystin-LREnzo Life SciencesCatalog #ALX-350-012
Protease Inhibitor Tablets cOmplete Mini EDTA free RocheCatalog #11836170001
Equipment:
- CO2 incubator for growing cells.
- Laminar flow hood for cell culture.
- Zeiss confocal laser scanning microscope.
Troubleshooting
Reverse transfection of HEK293 cells
Prepare a transfection mix by adding 0.6 µg of Flag-LRRK2 (or Flag-empty vector) cDNA and 1.8 µL of 1 mg/mL PEI solution into 150 µL of Opti-MEM for each well. Vortex for 00:00:20 /00:00:30 .
Note
Note:
- A total of 4 wells will be needed for each LRRK2 construct (see below), for which we recommend preparing a transfection mix with 2.7 µg of cDNA and 8.1 µg of PEI in 675 µL of Opti-MEM (enough for 4.5 wells).
- We recommend including a FLAG empty vector transfection to control for the specificity of LRRK2 staining in immunofluorescence imaging.
50s
Incubate the transfection mix for 00:20:00 at Room temperature to allow the DNA/PEI complex to form.
20m
For each LRRK2 construct, add 150 µL of the transfection mix to each well.
Note
Note: A total of 4 wells are needed for each LRRK2 construct: 2 wells for immunofluorescence microscopy performed in duplicate (Ibidi µ-plates), and 2 wells for immunoblotting analysis performed in duplicate (regular 24-well plates).
Remove culture medium from one flask of HEK293 cells.
Briefly rinse the cell layer with 0.25% (w/v) Trypsin - 0.53 millimolar (mM) EDTA solution to remove all traces of serum.
Add 2 mL of Trypsin-EDTA solution to the flask and incubate at 37 °C until the cell layer is dispersed.
Add 8 mL of complete growth medium and resuspend cells by gently pipetting.
Count the cells using the method of choice.
Resuspend the cells to a concentration of 8x104 cells per ml of complete growth medium.
Add 1 mL of cell suspension (8x104 cells) into the well containing the transfection mix.
Note
Note: If preparing many plates at once, we recommend not moving the plates for 10-15 min after adding the cell suspension to allow the cells to start attaching. This will help avoid cells swirling to the center of the well.
Transfer the plates to a humidified incubator maintaining 37 °C and 5% (v/v) CO2.
Sample preparation for immunoblotting analysis
45 hours after transfection, treat cells with 100 nanomolar (nM) MLI-2 or 0.1% (v/v) DMSO (control vehicle) and incubate for 03:00:00 at 37 °C in a humidified incubator maintaining 5% (v/v) CO2.
After treatment with MLi-2/DMSO, remove culture medium completely from each well using an aspirator.
Immediately add 50 µL of ice-cold complete lysis buffer to each well ensuring that the entire surface is covered by lysis buffer.
Transfer the plate On ice .
Scrape the cells on the dish using a cell lifter to ensure all cells are detached from the well.
Using a pipette, transfer the lysate to a 1.5 mL Eppendorf tube.
Leave samples On ice for 20/30 minutes to allow for efficient lysis.
Spin down lysates at 17000 x g, 4°C, 00:10:00 .
Transfer supernatant to a new Eppendorf tube and discard the pellet.
Proceed to quantitative immunoblotting analysis as described in dx.doi.org/10.17504/protocols.io.bsgrnbv6 (Quantitative Immunoblotting Analysis of LRRK2 Signalling Pathway).
Note
Note: Blot the samples for LRRK2 total and pS935 LRRK2 levels, Rab10 total and pT73 Rab10 levels, and tubulin or other loading control to assess LRRK2 expression levels and efficient LRRK2 inhibition in MLi-2 treated samples.
Sample preparation for immunofluorescence microscopy
45 hours after transfection, treat cells with 100 nanomolar (nM) MLI-2 or 0.1% (v/v) DMSO (control vehicle) and incubate for 03:00:00 at 37 °C in a humidified incubator maintaining 5% (v/v) CO2.
After treatment with MLi-2/DMSO, remove culture medium completely from each well using an aspirator.
Fix cells by adding 4% (v/v) PFA in PBS pre-warmed to 37 °C .
Incubate for 00:10:00 at Room temperature .
Remove PFA completely using a pipette and wash with PBS.
Note
Note: Samples can be kept in PBS at 4 °C for up to a week before proceeding to permeabilisation and staining.
Permeabilise cells by incubating with 1% (v/v) NP-40 alternative in PBS for 00:10:00 at Room temperature .
Remove the solution completely using an aspirator.
Block with 1% (w/v) BSA in PBS for 01:00:00 at Room temperature .
Prepare the primary antibody solution by diluting anti-Flag M2 antibody and anti-β-tubulin antibody in 0.2% (w/v) BSA in PBS (1:1000 and 1:500 dilution, respectively).
Incubate the samples with primary antibodies for 02:00:00 at 37 °C in a humidified chamber.
Note
Note: Incubation at 37 °C is necessary for the Flag antibody staining quality.
Wash the samples 3 times with 0.2% (w/v) BSA in PBS (10 minutes per wash).
Prepare the secondary antibody solution by diluting the secondary antibodies (Alexa Fluor 488 goat anti-mouse and Alexa Fluor 594 goat anti-rabbit) in 0.2% BSA in PBS (1:500 dilution). Add DAPI at 1 µL final concentration to the secondary antibody solution.
Incubate the samples with the secondary antibodies and DAPI at Room temperature for 01:00:00 in the dark.
Wash the samples 3 times with 0.2% (w/v) BSA in PBS (10 minutes per wash).
Note
Note: Samples can be kept in PBS at 4 °C for up to 2-3 weeks before proceeding to imaging.
Laser confocal imaging
Image cells using a Zeiss LSM 710 or 880 laser scanning microscopes using the x40 EC Plan-Neofluar (NA 1.3) objective with a zoom of 0.6 and optical section thickness of 1.0 mm (image size 2048x2048 pixels, pixel size 0.173 μm).
Image 50-100 cells from 4-6 randomly selected fields with Alexa Fluor 488-positive cells for each well (These are cells successfully transfected with Flag-LRRK2).
Cell counting and statistical analysis
Perform cell counting using the Image J Cell Counter feature.
Note
Note:
- Cell counting should be performed blinded to LRRK2 variant and treatment condition. For this purpose, randomised file names for the image files can be generated using a Python code script as detailed below (How to generate a Python code script to generate randomised file names).
- Cells can be divided into 3 categories based on their Alexa Fluor 488 signal (corresponding to LRRK2 staining):
1. cells showing filamentous/string-like staining (“filamentous”);
2. cells with no filamentous staining but containing punctate/aggresome-like staining (“punctate”);
3. cells with only cytosolic staining (“cytosolic”).
For each experimental condition, count cells in each category.
- DAPI and β-tubulin staining is used to ensure only cells containing a single nucleus are counted, avoiding cells that have not finished dividing or are multi-nuclear.
Once cell counting is done, data is unblinded and analysed as percentage of cells for each category using GraphPad Prism and applying a 2-way ANOVA and the post-hoc Dunnett’s test to evaluate statistical significance between different experimental conditions.
How to generate a Python code script to generate randomised file names
Copy the code below and change the folder directories according to where the data is located and where you would like the data with changed names to be located (the original data will not be changed).
import os
import string
import random
import shutil
def random_name(size=9, chars=string.ascii_uppercase + string.digits):
#This defines the random name as a string of 9 letters and digits.
return ''.join(random.choice(chars) for _ in range(size))
list=os.listdir("C:/Documents/Folder with the data/”)
#This is the folder containing the images.
for k in list: decode=open("decode.txt", "a")
#This creates a log of the original file names and new randomised file names so these can be decoded later.
codename=random_name()
print(codename)
decode.write(codename+".lsm"+'*'+k+'\n')
#Note: Change “.lsm” to whatever format you are using for the image files.
shutil.copy("C:/Documents/Folder with the data/"+k, "C:/Documents/Folder for the renamed data/"+codename+".lsm")
#The first folder is the folder containing the original image files (as before) and the second one is the folder for the renamed images to be copied to.
Select “Run” and the program will create the renamed copies for you to use.
Note
Note: This program was used via PyCharm 2017.3.3, before using it make sure you have Python installed (Python 3.6 was used, some changes might need to be made if using Python version 2.7).