Jun 23, 2025
IGGYPOP: Rapid and Large-Scale DNA Assembly Method
- Gony Dvir1,2,3,
- Zenan Xing1,2,3,
- Irina Beldman1,2,3,
- Andrés ivera1,2,3,4,
- Ian Wheeldon5,6,
- Sean Cutler1,2,3
- 1Center for Plant Cell Biology, University of California, Riverside, Riverside, CA, USA;
- 2Institute for Integrative Genome Biology, University of California, Riverside, Riverside, CA, USA;
- 3Botany and Plant Science, University of California, Riverside, Riverside, CA, USA;
- 4Centro de Ciencias Genómicas, Universidad Nacional Autónoma de México, MR, MX;
- 5Chemical and Environmental Engineering, University of California, Riverside, Riverside, CA, USA;
- 6Center for Industrial Biotechnology, University of California, Riverside, Riverside, CA, USA
- Gony Dvir: Equal contributions;
- Zenan Xing: Equal contributions;
- Sean Cutler: Corresponding author;



Protocol Citation: Gony Dvir, Zenan Xing, Irina Beldman, Andrés ivera, Ian Wheeldon, Sean Cutler 2025. IGGYPOP: Rapid and Large-Scale DNA Assembly Method. protocols.io https://dx.doi.org/10.17504/protocols.io.eq2lyqyzqvx9/v1
Manuscript citation:
Dvir G, Xing Z, Beldman I, Rivera A, Wheeldon I, Cutler SR. Synthesis of large single-transcript pathways from oligonucleotide pools: design of STARBURST, an autobioluminescent reporter (manuscript submitted)
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: May 24, 2025
Last Modified: June 23, 2025
Protocol Integer ID: 218895
Keywords: DNA Assembly, Nanopore Sequencing, IGGYPOP, indexed golden gate gene assembly, golden gate gene assembly from pcr, scale dna assembly method iggypop, genes from oligonucleotide pool, using gene, synthesizing gene, golden gate cloning, gene, oligonucleotide pool, iggypop, dna, barcoded amplicon
Funders Acknowledgements:
Defense Advanced Research Projects Agency CERES
Grant ID: D24AC00011-00
Abstract
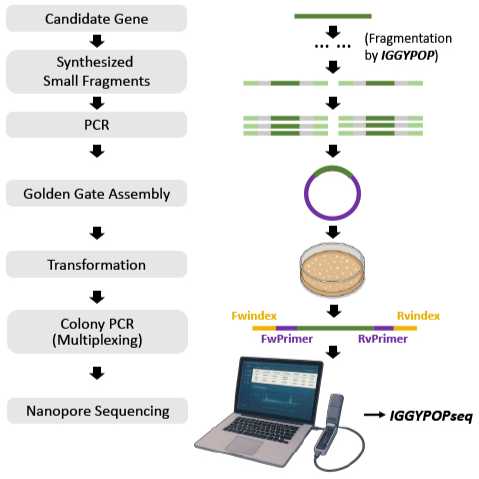
IGGYPOP (indexed golden gate gene assembly from PCR amplified oligonucleotide pools) is a pipeline for designing and synthesizing genes from oligonucleotide pools. Input sequences are fragmented into segments that can be amplified using gene-specific primers and reassembled by Golden Gate cloning. Sequence-verified constructs are then identified by nanopore sequencing of barcoded amplicons.
Protocol materials
GoTaq® Green Master Mix Promega CorparationCatalog #M7123
Invitrogen™ UltraPure™ BSA 50 mg/mlInvitrogenCatalog #AM2616
Nuclease-free WaterInvitrogen - Thermo FisherCatalog #AM9937
Flow Cell Wash KitOxford Nanopore TechnologiesCatalog #EXP-WSH004-XL
Ligation Sequencing Kit V14Oxford Nanopore TechnologiesCatalog #SQK-LSK114
NEBNext® Companion Module v2New England BiolabsCatalog #E7672
BbsI HFNew England BiolabsCatalog #R3539M
T4 DNA LigaseNew England BiolabsCatalog #M0202L
Phusion™ High-Fidelity DNA PolymeraseNew England BiolabsCatalog #M0530L
NEBridge Golden Gate Assembly Kit (BsmBI-v2)New England BiolabsCatalog #E1602L
Oligonucleotide Pool Design
Install iggypop
Detailed installation instructions, along with an overview of the available functions are provided on the official repository: https://github.com/cutlersr/iggypop/tree/main.
Generate oligo pool
Run iggypop, providing a file containing the sequences of interest as input and specifying a destination directory where the results will be saved. The default parameters will introduce synonymous mutations to remove internal BsaI and BsmBI sites, add external overhangs that allow cloning into the pPop vectors and BsaI sites to the 5' and 3' ends for subsequent Golden Gate cloning. You can adjust the parameters to suit the specifics of your experimental design via the command line or a .yml file. The yaml directory (https://github.com/cutlersr/iggypop/tree/main/yaml/) includes several preconfigured .yml files that provide the specific parameters for a variety of experiments. In addition to fasta formatted sequences, you can use Genbank formatted files as inputs; see the iggypop Readme file for details.
Retrieve results
Upon completion, several output files will be generated. Among these, the most important are: *_oligo_pool_to_order.fasta, which contains the designed oligo pool for synthesis and *__pcr_primers_required.fasta, which lists the gene-specific primers required for PCR amplification. Additionally, *_designed_seqs.fasta contains the assembled sequences. Verify these outputs before proceeding with the experimental workflows.
Oligo Amplification
Phusion PCR (96-Well Plate Format)
The fragments required for gene assembly are amplified from oligonucleotide pools using a set of reusable indexing (i.e., gene-specific) PCR primers; the primers required are located in the file iggypop/out/[project]/[project]_index_primers_required.fasta.
Template preparation:
Resuspend the oligo library in nuclease-free water to a final concentration of 1 ng/µL.
Prepare a working dilution by diluting 1:10 in nuclease-free water to achieve 100 pg/µL (0.1 ng/µL).
PCR Specifications (based on the Phusion protocol):
Phusion™ High-Fidelity DNA PolymeraseNew England BiolabsCatalog #M0530L
| Component | Volume (µL) | |
| Phusion Enzyme | 0.25 | |
| HF Buffer | 5 | |
| dNTPs (10 µM) | 0.5 | |
| Primer F+R mix (10 µM) | 5 | |
| Template (0.1 ng/µL) | 1 | |
| Nuclease-free water | to 25 |
Thermocycling Conditions:
| Step | Temperature | Time | |
| Initial Denaturation | 98°C | 30 seconds | |
| 30 Cycles | |||
| – Denaturation | 98°C | 10 seconds | |
| – Annealing | 60°C | 10 seconds | |
| – Extension | 72°C | 30 seconds | |
| Final Extension | 72°C | 5 minutes | |
| Hold | 12°C | Indefinite |
Quality Check
Run 3 µl on an agarose gel to assess amplification quality.
PCR Purification with Beads
To prepare your own beads, follow this protocol:
Protocol
CREATED BY
John B. Ridenour
Use 2× the PCR volume for purification (e.g., add 50 µL of beads to 25 µL PCR reaction).
Post-Purification QC
- Use Nanodrop to quantify a few samples and estimate the average DNA yield.
- Run 3 µL from each well of the 96-well plate on an agarose gel to check for missing or failed PCR reactions.
Golden Gate Assembly
One-step assembly -- Golden Gate Assembly (BsmBI)
Note
The default iggypop oligo design parameters are set up to work with pPlantPOP or pPOP plasmids, which use either BsmBI or BbsI with 5'-AATG/GCTT-3' overhangs on target genes for assemblies. If you need to use different overhangs and/or enzymes, update your iggypop parameters appropriately.
Using NEBridge-BsmBINEBridge Golden Gate Assembly Kit (BsmBI-v2)New England BiolabsCatalog #E1602L
- Reaction Setup (10 µL total):
| Component | Amount | |
| pPlantPOP or pPOP-BsmBI | 60 ng or 35 ng respectively | |
| Inserts (purified PCR product) | ~5.5 ng × average number of fragments | |
| 10X T4 DNA Ligase Buffer | 1 µL | |
| NEB Golden Gate Assembly Mix | 0.5 µL | |
| Nuclease-free water | to 10 µL |
2. Cycling Protocol:
(42°C, 5 min → 16°C, 5 min) × 90 cycles → 60°C, 5 min
Modification for 2-step assembly
Although assembly of long (>2.5 kb) sequences is possible, the assembly efficiency can be low, and identifying error-free clones often requires substantially more amplicon sequencing than for smaller targets. For longer sequences (>2 kb), we recommend using the two-step assembly mode, which breaks sequences into "step one" blocks assembled using BbsI and pPOP-BbsI. Sequence validated step one clones are identified, and the final genes are constructed in a second step using pPlantPOP (w/ BsmBI). You need to use the 2-step YAML files for oligo pool design.
Golden Gate Assembly -- first step is into pPlantPOP w/ BbsI
BbsI HFNew England BiolabsCatalog #R3539M
T4 DNA LigaseNew England BiolabsCatalog #M0202L
- Reaction Setup (10 µL total):
| Component | Amount | |
| pPOP-BbsI | 35 ng | |
| Inserts (purified PCR product) | ~5.5 ng × average number of fragments | |
| 10X T4 DNA Ligase Buffer | 1 µL | |
| BbsI | 0.5 µL | |
| T4 DNA ligase | 0.5 µL | |
| Nuclease-free water | to 10 µL |
2. Cycling Protocol:
(37°C, 5 min → 16°C, 5 min) × 90 cycles → 60°C, 5 min
3. See step 22 for assembly of the final clone after obtaining sequence-verified step 1 clones.
Transformation
- Prepare chemically competent E. coli using the CCMB80 method; freeze in ~ 5 mL aliquots.
- Thaw 5 mL competent cells on ice, dispense 50 µL per well of a 96-well plate on ice
- Add 2 µL of the assembly reaction per well.
- Incubate on ice for 30 minutes.
- Perform heat shock at 42°C for 1 min in a thermal cycler.
- Transfer to a 2 mL 96-well plate containing 250 µL SOC medium per well.
- Incubate at 37°C with shaking for 1 hour.
- Centrifuge at 1000 RPM for 5 minutes, remove 200 µL SOC, and shake again for at least 30 minutes.
- Plate the entire suspension on 9 cm LB agar plates supplemented with 25 mg/L chloramphenicol (pPOP-BsmBI and pPOP-BbsI ) or 100 mg/L spectinomycin (pPlantPOP)
Barcoded Amplicons Generation
Colony PCR with Barcoded Primers
- The primers we use for generating barcoded amplicons with pPOP or pPlantPOP are provided here. After synthesizing them, they are arrayed combinatorially (https://github.com/cutlerlab/Construct-Validation-for-IGGYPOPseq/tree/main/Input/PrimerAssignment) in 96-well PCR plates (10 µM, see 10.2) and used for colony PCR.
Colony Preparation
- Aim to screen 6 to 8 colonies per construct.
- Prepare 96-well plates with 20 µL ddH2O per well.
- Pick colonies and resuspend in water to use in PCR reactions
- Spot colony suspensions onto single-well rectangular LB + antibiotic plates to store the bacterial stocks for recovery after sequence verification.
Primer Preparation:
Barcoding primers are arrayed into 96-well plates that can be used for multiple experiments
- Dilute barcoded primers to 1 µM in a new plate.
- Mix the forward and reverse primers in separate plates following the plate layout for the specific plasmid provided here.
PCR Setup (10 µL total):
GoTaq® Green Master Mix Promega CorparationCatalog #M7123
| Component | Volume (µL) | |
| GoTaq Green Master Mix | 5 | |
| Primer F+R mix (1 µM) | 2 | |
| Colony suspension | 1 | |
| Nuclease-free water | to 10 |
Stocking Colonies:
- Use a multi-channel set at 3 µl pipette, pipette ~2 µL drops of the suspension onto the LB plate in an organized pattern.
- Add the remaining ~1 µL to the PCR reaction.
- Seal PCR plates tightly and mix by tapping.
- Spin the plates down.
- Cycling Conditions:
| Step | Temperature | Time | |
| Initial Denaturation | 94°C | 1.5 mins | |
| 30 Cycles | |||
| – Denaturation | 94°C | 10 seconds | |
| – Annealing | 55°C | 10 seconds | |
| – Extension | 72°C | 60–120 sec (depending on amplicon length) | |
| Final Extension | 72°C | 5 minutes | |
| Hold | 12°C | Indefinite |
Quality Check
Run 3 µl on an agarose gel to assess amplification quality.
PCR Product Purification
- Pool 2 µL from each well into a single tube for each plate separately.
- Purify 25 µL from each pooled tube using a 1× volume of magnetic beads using standard bead purification protocol.
- Elute in 15 µL.
- Pool eluates to achieve even representation across the samples, targeting ~1 µg total DNA.
Nanopore Sequencing
Required equipment and reagents.
- Reagents:
Ligation Sequencing Kit V14Oxford Nanopore TechnologiesCatalog #SQK-LSK114
NEBNext® Companion Module v2New England BiolabsCatalog #E7672
Invitrogen™ UltraPure™ BSA 50 mg/mlInvitrogenCatalog #AM2616
Nuclease-free WaterInvitrogen - Thermo FisherCatalog #AM9937
Flow Cell Wash KitOxford Nanopore TechnologiesCatalog #EXP-WSH004-XL
- Equipment:
Check the flow cell.
Library preparation.
Priming and loading the MinION flow cell.
Data acquisition and basecalling.
Note
Important Sequencing Settings:
- Basecalling: High-accuracy basecalling by Dorado
- Output file format: pod5 & fastq
Wash and store the flow cell.
Data Analysis
Create a project workspace.
- Make a new directory for this run (e.g., IGGYPOP_YYYYMMDD).
- Inside it, create one subfolder named Input, for the files you will prepare below.
Generate SampleInfo.tsv.
- Alternatively, you may open a spreadsheet editor and enter one row per sample with the required columns, saving it as SampleInfo.tsv (tab-separated).
| Column | Type | |
| primer_index | string | |
| SampleID | string | |
| n_frags | int | |
| CDS_length (Optional) | int | |
| ReferenceName | string | |
| ReferenceSequence | string | |
| FwBarcode | string | |
| FwPrimer | string | |
| RvBarcode | string | |
| RvPrimer | string |
The detailed description of each column can be found in our GitHub repo (https://github.com/cutlerlab/Construct-Validation-for-IGGYPOPseq.git).
- Move the file into the Input/ folder.
Generate passed_all.fastq.
- Locate the base-called reads folder (usually fastq_pass/).
- Unzip any .gz files (skip if already in plain .fastq format), then concatenate all passed read files into one file named passed_all.fastq. After that, move the concatenated file into the Input/ folder. You can use the following command to accomplish this.
zcat fastq_pass/*.fastq.gz > Input/passed_all.fastq
Run the IGGYPOPseq pipeline.
- Choose the execution environment and follow the corresponding instructions on our GitHub repo (https://github.com/cutlerlab/Construct-Validation-for-IGGYPOPseq.git).
| Environment | Link to Instructions | |
| Linux | https://github.com/cutlerlab/Construct-Validation-for-IGGYPOPseq?tab=readme-ov-file#--linux | |
| Docker | https://github.com/cutlerlab/Construct-Validation-for-IGGYPOPseq?tab=readme-ov-file#--docker | |
| Compute cluster | https://github.com/cutlerlab/Construct-Validation-for-IGGYPOPseq?tab=readme-ov-file#--parallelized-analyses-with-slurm |
Check and store the analysis results.
- All the results are available in the following directory: IGGYPOP_YYYYMMDD/Analysis_Results/.
- Store the folder in your institutional or long-term repository.
2nd assembly step for sequence-verified step 1 clones
Golden Gate Assembly Using NEBridge-BsmBI into pPlantPOP
NEBridge Golden Gate Assembly Kit (BsmBI-v2)New England BiolabsCatalog #E1602L
Reaction Setup (10 µL total):
| Component | Amount | |
| pPlantPOP | 60 | |
| Inserts (sequence validated pPOP plasmids) | ~30 ng each | |
| 10X T4 DNA Ligase Buffer | 1.5 µL | |
| NEB Golden Gate Assembly Mix | 0.5 µL | |
| Nuclease-free water | to 15 µL |
Cycling Protocol:
(42°C, 5 min → 16°C, 5 min) × 90 cycles → 60°C, 5 min
- for the transformation procedure.
- Plate cells on spectinomycin (100 mg/L).
- Prep 1 - 2 colonies per clone, digest for initial confirmation; whole-plasmid sequence for final validation.
Protocol references
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, et al (2021) Twelve years of SAMtools and BCFtools. Gigascience. doi: 10.1093/gigascience/giab008
De Coster W, Rademakers R (2023) NanoPack2: population-scale evaluation of long-read sequencing data. Bioinformatics 39: btad311
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797
Krehenwinkel H, Pomerantz A, Henderson JB, Kennedy SR, Lim JY, Swamy V, Shoobridge JD, Graham N, Patel NH, Gillespie RG, et al (2019) Nanopore sequencing of long ribosomal DNA amplicons enables portable and simple biodiversity assessments with high phylogenetic resolution across broad taxonomic scale. Gigascience 8: giz006
Li H (2018) Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100
Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842
Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, Manke T (2016) deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44: W160–5
Rice P, Longden I, Bleasby A (2000) EMBOSS: The European molecular biology open software suite. Trends Genet 16: 276–277
Tange O (2021) GNU Parallel 20220122 ('20 years’). doi: 10.5281/zenodo.5893336