Feb 19, 2026
Version 1
Generation of cirVDJseq libraries from 3’-barcoded cDNA V.1
- Izabela Plumbom1,2,
- Benedikt Obermayer3,
- Thomas Conrad1,2
- 1Max Delbrück Center for Molecular Medicine in the Helmholtz Association, Berlin Germany;
- 2Berlin Institute of Health (BIH) at Charité - Universitätsmedizin Berlin , Germany;
- 3Berlin Institute of Health (BIH) at Charité - Universitätsmedizin Berlin, Germany



External link: https://www.biorxiv.org/content/10.1101/2025.09.16.675546v1
Protocol Citation: Izabela Plumbom, Benedikt Obermayer, Thomas Conrad 2026. Generation of cirVDJseq libraries from 3’-barcoded cDNA. protocols.io https://dx.doi.org/10.17504/protocols.io.q26g77zx1gwz/v1
Manuscript citation:
Plumbom I et al. circVDJ-seq for T cell clonotype detection in single-cell and spatial multi-omics. bioRxiv 2025. doi:10.1101/2025.09.16.675546
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: December 17, 2025
Last Modified: February 19, 2026
Protocol Integer ID: 235255
Keywords: circVDJ-seq, T cell receptor, TCR sequencing, VDJ profiling, immune repertoire, T cell clonality, 3’-barcoded cDNA, single-nucleus RNA-seq, RNA+ATAC multi-omics, spatial transcriptomics, circular DNA library preparation, Gibson assembly, 10x Genomics, human tissue, adaptive immunity, cancer immunology, infectious diseases, COVID-19, 10x genomics vdj library construction, robust recovery of tcrα, generation of cirvdjseq library, cirvdjseq library, efficient tcr vdj profiling, ready tcr library, immune microenvironments across diverse clinical sample, spatial transcriptomics workflow, tapestation high sensitivity dna assay, nucleus nucleus rna, cdna, tcrα, diverse clinical sample, circvdj, clonal repertoire, remaining linear dna, linear dna, immune microenvironment, quality vdj library peak
Abstract
This protocol describes circVDJ-seq, a method for simplified and cost-efficient TCR VDJ profiling from 3’-barcoded cDNA generated in single-cell or single-nucleus nucleus RNA-seq, RNA+ATAC multi-omics, or spatial transcriptomics workflows. 3’-barcoded cDNA is modified with Gibson assembly overhangs, circularized, depleted of remaining linear DNA, and subjected to nested PCR and 10x Genomics VDJ library construction to generate sequencing-ready TCR libraries.
The expected outcome is a discrete, high-quality VDJ library peak on TapeStation High Sensitivity DNA assays with sufficient yield for Illumina sequencing using custom primers. When applied to human tissues, circVDJ-seq enables robust recovery of TCRα/β VDJ sequences and clonal repertoires, allowing characterization of T cell clonality and immune microenvironments across diverse clinical samples.


Image Attribution
Image reproduced from Plumbom et al., "circVDJ-seq for T cell clonotype detection in single-cell and spatial multi-omics", bioRxiv 2025. doi:10.1101/2025.09.16.675546
Guidelines
- This protocol is optimized for TCR V(D)J enrichment from 3’-barcoded cDNA derived from single-cell or single-nucleus RNA-seq, RNA+ATAC, or spatial transcriptomics workflows. Using other cDNA types may require re-optimization of input amount and cycle numbers.
- Whenever possible, start with high-quality 3’-barcoded cDNA that has already passed QC (TapeStation/Fragment Analyzer and Qubit). Degraded or very low-complexity input will reduce clonotype recovery.
- Maintain accurate AMPure XP/SPRIselect bead ratios (0.8X–0.9X as indicated). Deviations in bead volume critically affect size selection and yield. Mix beads thoroughly before use.
- Avoid over-drying bead pellets. Over-dried beads are difficult to resuspend and can cause large losses of DNA. Pellets should appear matte but not cracked.
- Use freshly prepared 80% ethanol for washes. Residual salts or lower ethanol concentration will impair cleanup efficiency.
- Adjust the number of PCR cycles (especially V(D)J Amplification 1) according to sample type and expected T-cell content. Over-amplification can introduce bias and increase PCR artifacts.
- Perform QC after each major stage (post-PCR#1, post-circularization, post-V(D)J enrichment, final libraries) using Qubit and TapeStation to monitor yield and fragment size and to identify issues early.
- For library construction, follow the Chromium Next GEM Single Cell 5’ v2 (Dual Index) V(D)J Library Construction instructions closely, using the custom V(D)J cDNA as input and the custom sequencing primers listed in the protocol.
- Include appropriate controls where possible (e.g. T-cell–rich reference sample or a previously validated sample) to benchmark clonotype recovery and library performance between runs.
Materials
Chemicals/kits
| Chemicals/Kits | Additional Description | Manufacturer | |
| Ethanol | Catalog number: 11096.02 | SERVA Electrophoresis | |
| Elution Buffer (EB) | Catalog number: 19086 | QIAGEN | |
| Agencourt AMPure XP | REF: A63881 | Beckman Coulter GmbH | |
| KAPA HiFi HotStart ReadyMixPCR Kit | REF: 07958927001 | KAPA Biosystems | |
| QubitTM dsDNA HS Assay Kit | Quantitation Range: 0.2-100 ng | Invitrogen | |
| High Sensitivity D1000 ScreenTape Kit | Sizing Range: 35 – 1000 bp | Agilent Technologies | |
| High Sensitivity D5000 ScreenTape Kit | Sizing Range: 100 – 5000 bp | Agilent Technologies | |
| Water | Bioperformance certified, Lot No. RNBJ7736 | Sigma-Aldrich | |
| NEBuilder HiFi DNA Assembly Master Mix | Catalog number: E261S | New England BioLabs | |
| CutSmart Buffer | Catalog number: B7204S | New England BioLabs | |
| Lambda Exonuclease | Catalog number: M0262S, 5,000 units/mL | New England BioLabs | |
| Library Construction Kit | Lot: 160026, PN: 1000190 | 10X Genomics | |
| KAPA Library Quantification (Illumina) Primers & LightCycler 480 qPCRMix | Lot:0000121161 | Roche | |
| KAPA Library Quantification (Illumina) DNA Standards 1-6 | Lot: 0000120225 | Roche | |
| Tween-20 | Lot: P9416 | Sigma-Aldrich |
Table 1.1: Overview of chemicals/kits
Devices and Software
| Device/Software | Model/Designation | Manufacturer | |
| Vortex Mixer | Vortex-Genie 2 | Scientific Industries | |
| PCR Thermal Cyclers | Mastercycler X50s | eppendorf | |
| Thermomixer | with thermoblocks for 24 reaction vessels 1.5 mL, 2 mL | eppendorf | |
| Magnetic Stand 0,2 | to 8 purifications in parallel | Thermo Fisher Scientific | |
| Thermoblock | ThermoStat plus | eppendorf | |
| Fluorometer | Qubit 3 Fluorometer | Thermo Fisher Scientific | |
| Mini-centrifuge | Rotilabo‱-mini-centrifuge "Uni-fuge" | Carl Roth | |
| Centrifuge | Centrifuge 5427 R | eppendorf | |
| PCR Plate Spinner | max. capacity: 2 plates | VWR International | |
| Repetitive pipette | Multipette‱ E3x | eppendorf | |
| Charging Stand | Charging Stand 2 for one electronic Multipette | eppendorf | |
| Dispenser tips | Eppendorf Combitips advanced (0,1 ml, 0,5 ml, 2,5 ml) | eppendorf | |
| Pipette Tips | xTip 4, low retention manual filter pipette tip (20 μL, 200 μL) | Biotix | |
| DNA LoBind Tubes | 1,5 mL, 0,5 mL | eppendorf | |
| Thin-walled Tubes with Flat Caps | 0.5 mL | Thermo Fisher Scientific | |
| Pipette tips | SafeSeal-Tips Professional Line (10μL, 20 μL, 200μL, 1000μL) | Biozym Scientific | |
| Pipettes | Eppendorf Research plus pipette (0.1–2.5μL, 0.5–10 μL, 2–20μL, 10–100 μL, 20–200μL, 100–1,000 μL) | eppendorf | |
| Multichannel pipettes | Pipet-Life XLS, 8-Channels (0.5-10 μL, 2-20 μL, 20-200 μL) | RAININ | |
| PCR 8er-SoftStrips | 0.2 ml | Biozym Scientific | |
| PCR Tubes | PCR-02-L-C, 0.2mL maximum recovery, thin wall, clear | Axygen | |
| Pipettor | Pipetboy acu 2 | INTEGRA Biosciences | |
| Serological pipettes | 5 mL, 10 mL | Sarstedt | |
| Conical Centrifuge Tubes | FalconTM, 15mL, 50mL | Fisher Scientific | |
| PCR plates for Roche LightCycler 480 | Catalog number: 732-1462, No. Of wells: 96 | VWR International | |
| PCR Seal Sheets | Optically clear film, adhesive seal. 140x77mm | 4titude | |
| LightCycler 480 Sealing Foil | REF: 04729757001 | Roche Molecular System | |
| Optical Film Compression Pad | 4TI-0563 | 4titude | |
| Benchtop Cooler | StrataCooler LP | Agilent Technologies | |
| Vortex Mixer | IKA MS3 Vortexer | IKA | |
| LightCycler 480 II System | Serial number: 27785 | Roche | |
| TapeStation | Agilent 4200 TapeStation System | Agilent Technologies | |
| TapeStation Analysis Software | - | Agilent Technologies |
Table 1.2: Devices and softwares
Primer
| A | B | C | D | |
| Oligo name | Sequence (5’ -> 3’) | Comment | Manufacturer | |
| TCRGOT_1 | GAGCAAGTATGTACCGTTCCAAGCAGTGGTATCAACGCAGAG | IDT | ||
| TCRGOT_2 | GGAACGGTACATACTTGCTCCTACACGACGCTCTTCCGATCT | IDT | ||
| TRAC_3UTR_1 | /5BiotinTEG/GTCTGGGCGTGTTGTATGTC | 5′-BiTEG modification | IDT | |
| TRAC_3UTR_2 | /5BiotinTEG/GTGTTGTATGTCCTGCTGCC | 5′-BiTEG modification | IDT | |
| TRBC1_3UTR | /5BiotinTEG/CACACTCACGGCTGAAATCT | 5′-BiTEG modification | IDT | |
| TRBC2_3UTR | /5BiotinTEG/CCCTGAAGATTGAGCTCCCA | 5′-BiTEG modification | IDT | |
| HTCR_o_alpha | /5BiotinTEG/TGAAGGCGTTTGCACATGCA | 5′-BiTEG modification | IDT | |
| HTCR_o_beta | /5BiotinTEG/TCAGGCAGTATCTGGAGTCATTGAG | 5′-BiTEG modification | IDT | |
| HTCR_i_alpha | /5BiotinTEG/AGTCTCTCAGCTGGTACACG | 5′-BiTEG modification | IDT | |
| HTCR_i_beta | /5BiotinTEG/TCTGATGGCTCAAACACAGC | 5′-BiTEG modification | IDT | |
| P5_SI_TT | AATGATACGGCGACCACCGAGATCTACAC NNNNNNNNNN GTGACTGGAGTTCAGACGTG*T | IDT | ||
| P5_SI-TT-A1 | AATGATACGGCGACCACCGAGATCTACAC GTAACATGCG GTGACTGGAGTTCAGACGTG*T | IDT | ||
| P5_SI-TT-A2 | AATGATACGGCGACCACCGAGATCTACAC GTGGATCAAA GTGACTGGAGTTCAGACGTG*T | IDT | ||
| P5_SI-TT-A3 | AATGATACGGCGACCACCGAGATCTACAC CACTACGAAA GTGACTGGAGTTCAGACGTG*T | IDT | ||
| P5_SI-TT-A4 | AATGATACGGCGACCACCGAGATCTACAC CTCTAGCGAG GTGACTGGAGTTCAGACGTG*T | IDT | ||
| P5_SI-TT-A5 | AATGATACGGCGACCACCGAGATCTACAC GTAGCCCTGT GTGACTGGAGTTCAGACGTG*T | IDT | ||
| P5_SI-TT-A6 | AATGATACGGCGACCACCGAGATCTACAC TAACGCGTGA GTGACTGGAGTTCAGACGTG*T | IDT | ||
| P5_SI-TT-A7 | AATGATACGGCGACCACCGAGATCTACAC TCCCAAGGGT GTGACTGGAGTTCAGACGTG*T | IDT | ||
| P5_SI-TT-A8 | AATGATACGGCGACCACCGAGATCTACAC CGAAGTATAC GTGACTGGAGTTCAGACGTG*T | IDT | ||
| P5_SI-TT-A9 | AATGATACGGCGACCACCGAGATCTACAC AAGTGGAGAG GTGACTGGAGTTCAGACGTG*T | IDT | ||
| P5_SI-TT-A10 | AATGATACGGCGACCACCGAGATCTACAC CGTGACATGC GTGACTGGAGTTCAGACGTG*T | IDT | ||
| P7_TRAC_3UTR_f3_i7 | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGCGNNNNNNNNNNGTATGTCCTGCTGCCGATGC*C | IDT | ||
| P7_TRBC1_3UTR_f_i7 | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGCGNNNNNNNNNNACGGCTGAAATCTCCCTAACCCA*G | IDT | ||
| P7_TRBC2_3UTR_f_i7 | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGCGNNNNNNNNNNGATTGAGCTCCCAACCCCCAA*G | IDT | ||
| P7_TRAC_3UTR_f3_i7_A1 | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGCG AGTGTTACCT GTATGTCCTGCTGCCGATGC*C | IDT | ||
| P7_TRBC1_3UTR_f_i7_A1 | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGCG AGTGTTACCT ACGGCTGAAATCTCCCTAACCCA*G | IDT | ||
| P7_TRBC2_3UTR_f_i7_A1 | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGCG AGTGTTACCT GATTGAGCTCCCAACCCCCAA*G | IDT | ||
| P7_TRAC_3UTR_f3_i7_A2 | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGCG GCCAACCCTG GTATGTCCTGCTGCCGATGC*C | IDT | ||
| P7_TRBC1_3UTR_f_i7_A2 | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGCG GCCAACCCTG ACGGCTGAAATCTCCCTAACCCA*G | IDT | ||
| P7_TRBC2_3UTR_f_i7_A2 | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGCG GCCAACCCTG GATTGAGCTCCCAACCCCCAA*G | IDT | ||
| P7_TRAC_3UTR_f3_i7_A3 | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGCG TTAGACTGAT GTATGTCCTGCTGCCGATGC*C | IDT | ||
| P7_TRBC1_3UTR_f_i7_A3 | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGCG TTAGACTGAT ACGGCTGAAATCTCCCTAACCCA*G | IDT | ||
| P7_TRBC2_3UTR_f_i7_A3 | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGCG TTAGACTGAT GATTGAGCTCCCAACCCCCAA*G | IDT | ||
| P7_TRAC_3UTR_f3_i7_A4 | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGCG TATCTTCATC GTATGTCCTGCTGCCGATGC*C | IDT | ||
| P7_TRBC1_3UTR_f_i7_A4 | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGCG TATCTTCATC ACGGCTGAAATCTCCCTAACCCA*G | IDT | ||
| P7_TRBC2_3UTR_f_i7_A4 | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGCG TATCTTCATC GATTGAGCTCCCAACCCCCAA*G | IDT | ||
| P7_TRAC_3UTR_f3_i7_A5 | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGCG GAGCATCTAT GTATGTCCTGCTGCCGATGC*C | IDT | ||
| P7_TRBC1_3UTR_f_i7_A5 | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGCG GAGCATCTAT ACGGCTGAAATCTCCCTAACCCA*G | IDT | ||
| P7_TRBC2_3UTR_f_i7_A5 | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGCG GAGCATCTAT GATTGAGCTCCCAACCCCCAA*G | IDT | ||
| P7_TRAC_3UTR_f3_i7_A6 | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGCG CCCTAACTTC GTATGTCCTGCTGCCGATGC*C | IDT | ||
| P7_TRBC1_3UTR_f_i7_A6 | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGCG CCCTAACTTC ACGGCTGAAATCTCCCTAACCCA*G | IDT | ||
| P7_TRBC2_3UTR_f_i7_A6 | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGCG CCCTAACTTC GATTGAGCTCCCAACCCCCAA*G | IDT | ||
| P7_TRAC_3UTR_f3_i7_A7 | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGCG TACTACCTTT GTATGTCCTGCTGCCGATGC*C | IDT | ||
| P7_TRBC1_3UTR_f_i7_A7 | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGCG TACTACCTTT ACGGCTGAAATCTCCCTAACCCA*G | IDT | ||
| P7_TRBC2_3UTR_f_i7_A7 | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGCG TACTACCTTT GATTGAGCTCCCAACCCCCAA*G | IDT | ||
| Sequencing primers: | readout: | |||
| spTCR_Read1 (90) | GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | VDJ sequence | IDT | |
| spTCR_Read2 (28) | CATACTTGCTCCTACACGACGCTCTTCCGATCT | UMI and cell barcode | IDT | |
| spTCR_Read3 (10) | AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC | i7 Index | IDT | |
| spTCR_Read4 (10) | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGC*G | i5 Index | IDT |
Table 1.3: Overview of the primers
Troubleshooting
Before start
- Ensure that 3’-barcoded cDNA from the upstream single-cell / single-nucleus / spatial workflow is available, quantified, and stored at −20 °C or −80 °C.
- Order and resuspend all primers and oligos used in the protocol (TCRGOT primers, TRAC/TRBC primers, outer/inner TCR primers, indexing primers, custom sequencing primers). Verify sequences and working concentrations.
- Thaw and prepare all buffers and enzymes (KAPA HiFi HotStart ReadyMix, NEBuilder HiFi DNA Assembly Master Mix, Lambda exonuclease + buffer, AMPure XP beads, SPRIselect, 10x Genomics V(D)J reagents). Mix thoroughly and keep enzymes on ice.
- Bring AMPure XP and SPRIselect beads to room temperature for at least 30 minutes and vortex well to ensure homogeneous suspension before use.
- Pre-program thermal cyclers with all required PCR and incubation profiles (PCR#1, circularization, Lambda exonuclease digestion, V(D)J Amplification 1 & 2, fragmentation/A-tailing, adapter ligation, indexing PCR).
- Verify access to all required equipment:
- Magnetic rack compatible with the tube/plate format used
- Qubit (or equivalent) with dsDNA HS assay kit
- TapeStation (or equivalent) with HS D5000 and D1000 ScreenTapes
- Refrigerated microcentrifuge
- Thermomixer or heating block with shaking option
- Illumina NextSeq 550 (or access to a facility that provides runs with the 90–28–10–10 configuration).
- Prepare a clean PCR work area with filtered pipette tips, dedicated pipettes.
- Plan sample layout, indices, and multiplexing strategy in advance to ensure non-overlapping sample indices in the sequencing run.
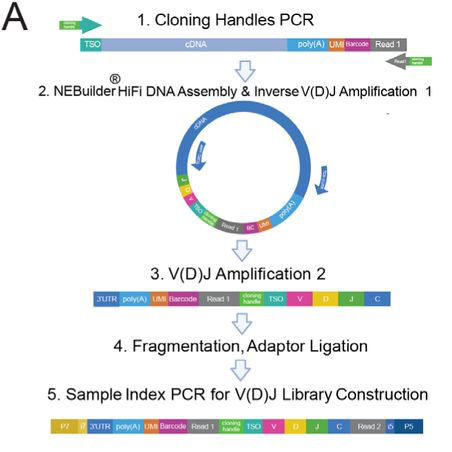
PCR #1 – amplification of 3’-barcoded cDNA
This step amplifies the 3’-barcoded cDNA and adds the Gibson assembly overhangs required for circularization.
Input: 1–15 ng of 3’-barcoded cDNA
Prepare PCR #1 reaction mix on ice
For each 25 μL reaction:
| Component | 25 μL reaction | Final conc. | |
| PCR-grade water | Up to 25 μL | N/A | |
| 2X KAPA HiFi HotStart ReadyMix | 12.5 μL | 1X | |
| 10 μM TCRGOT_1 primer | 0.75 μL | 0.3 μM | |
| 10 μM TCRGOT_2 primer | 0.75 μL | 0.3 μM | |
| 3’-barcoded cDNA template | 1-15 ng | 1-15 ng |
PCR#1 reaction mix
- Assemble all components on ice.
- Mix gently by pipetting and briefly spin down to collect liquid at the bottom.
PCR cycling
Place the tubes in a pre-cooled thermal cycler and run the following program (6 cycles of steps 3-5):
| A | B | C | D | |
| Step | Temperature | Duration | Cycles | |
| Initial denaturation | 95 °C | 3 min | 1 | |
| Denaturation | 98 °C | 20 s | 6 | |
| Annealing | 65 °C | 30 s | ||
| Extension | 72 °C | 2 min | ||
| Final extension | 72 °C | 5 min | 1 |
PCR#1 programm design
- Hold at 4 °C until proceeding to cleanup.
PCR #1 Cleanup
This cleanup removes primers, enzymes, and small fragments using AMPure XP beads.
- Bring the AMPure XP beads to the room temperature for at least 30 minutes and thoroughly vortex the to fully resuspend.
- Add 20 μL AMPure XP beads (0.9X) to each 25 μL PCR reaction.
- Mix by pipetting up and down ~15 times, or vortex.
- Incubate for 5 min at room temperature to allow DNA to bind to the beads.
- Place the tubes on a magnetic rack (high) for 5minutes or until the solution becomes clear and the beads are fully pelleted.
- Carefully remove and discard the supernatant without disturbing the bead pellet.
- Add 200 μL of freshly prepared 80% ethanol to the pellet and incubate for ~30 s.
- Remove the ethanol carefully.
- Repeat the ethanol wash (steps 7–8) for a total of 2 washes.
- Air-dry the bead pellet for 2-3 min (do not overdry; pellets should appear matte but not cracked).
- Remove the tubes from the magnet and add 20.5 μL EB buffer.
- Resuspend the beads thoroughly by pipetting up and down ~15 times, or vortex.
- Incubate for 2 min at room temperature to elute DNA.
- Place the tubes back on the magnet (low) until the solution clears.
- Transfer 20 μL of the clear supernatant to new tubes. This is your cleaned PCR #1 product.
PCR #1 Quality Control
Assess yield and fragment size distribution:
- Qubit dsDNA HS assay – to quantify DNA concentration.
- Agilent TapeStation HS D5000 – to confirm expected fragment size and absence of primer-dimers.
Circularization of cDNA
In this step, Gibson assembly is used to circularize the amplified cDNA.
Prepare circularization reaction
For each sample:
| Component | Volume | |
| cDNA template | 15–20 ng | |
| 1X CutSmart buffer | to 170 μL total | |
| 2X NEBuilder HiFi DNA Assembly Master Mix | 10 μL | |
circularization reaction mix
- Combine cDNA, CutSmart buffer, and NEBuilder HiFi DNA Assembly Master Mix in a total reaction volume of 200 μL.
- Mix thoroughly by pipetting and briefly spin down.
- Incubate at 50 °C for 1 hour in a thermomixer or thermal cycler (optional gentle shaking).
- After incubation, proceed immediately to the next step.
Circularization Cleanup
Purify the circularized cDNA using AMPure XP beads.
- Vortex (brought to the room temperature for at least 30min) AMPure XP beads to completely resuspend.
- Add 0.9X volume AMPure XP beads to each circularization reaction.
- Mix by pipetting 15 times.
- Incubate 5 min at room temperature.
- Place tubes on the magnet (high) until the solution is clear and beads are pelleted.
- Carefully remove and discard the supernatant.
- Add 200 μL 80% ethanol to the pellet and incubate ~30 s.
- Remove the ethanol carefully.
- Repeat the ethanol wash (steps 7–8) for a total of 2 washes.
- Air-dry the pellet for 2-3 min (avoid overdrying).
- Remove tubes from the magnet and add 20.5 μL EB buffer.
- Resuspend the beads thoroughly (pipette up and down ~15 times).
- Incubate 5 min at room temperature.
- Place tubes on the magnet (low) until the solution clears.
- Transfer 20 μL of the eluate to new tubes. This is the circularized cDNA.
Lambda Exonuclease Digestion
This step removes residual linear DNA, enriching for circular molecules.
Prepare the following mix for each sample:
| Component | Volume | |
| Circularized DNA | 20 μL | |
| 10X Lambda Exonuclease Buffer | 5 μL | |
| Lambda Exonuclease Enzyme | 1 μL | |
| Nuclease-free H₂O | 24 μL | |
| Total | 50 μL |
Lambda Exonuclease Digestion Reaction Mix
a. Incubation
- Incubate at 37 °C for 30 min to digest remaining linear DNA.
b. Enzyme inactivation
- Stop reaction by adding EDTA to 10mM.
- Heat-inactivate at 75 °C for 10 min.
- Cool samples on ice or at 4 °C before proceeding to cleanup.
Post-digestion Cleanup
Purify the circularized, exonuclease-treated DNA using AMPure XP beads.
- Bring the AMPure XP beads to the room temperature for at least 30 minutes and thoroughly vortex the to fully resuspend.
- Add 0.8X volume AMPure XP beads to each 50 μL digestion reaction.
- Mix by pipetting ~15 times.
- Incubate 5 min at room temperature.
- Place tubes on the magnet until the solution is clear (~5 min).
- Remove the supernatant carefully.
- Add 80% ethanol to wash the beads (originally written as 2 mL; please confirm – other washes use 200 μL).
- Wait 30 s, then remove the ethanol.
- Repeat the ethanol wash for a total of 2 washes.
- Air-dry the pellet for 2-3 min.
- Remove tubes from the magnet and add 25.5 μL EB + 0.05% Tween-20.
- Resuspend beads thoroughly by pipetting ~15 times.
- Incubate 5 min at room temperature.
- Place tubes on the magnet (low) until the solution clears.
- Transfer 25 μL of eluate to new tubes. This is your circularized cDNA ready for V(D)J amplification.
Circularization QC
Evaluate DNA yield and fragment size:
- Qubit dsDNA HS assay
- Agilent TapeStation HS D5000
V(D)J Amplification 1 (outer PCR)
This nested PCR enriches TCR α and β V(D)J regions from circularized cDNA.
a. Prepare V(D)J 1 primer mix on ice
| Primer | Volume | |
| 100 μM TRAC_3UTR_1 | 1 μL | |
| 100 μM HTCR_o_alpha | 1 μL | |
| 100 μM TRBC1_3UTR | 1 μL | |
| 100 μM TRBC2_3UTR | 1 μL | |
| 100 μM HTCR_o_beta | 1 μL | |
| H₂O | 5 μL | |
| Total | 10 μL |
V(D)J primer mix
b. Prepare V(D)J Amplification 1 reaction mix on ice
| Component | Volume | Final conc. | |
| cDNA template | 22.5 μL | — | |
| 2X KAPA HiFi HotStart ReadyMix | 25.6 μL | 1X | |
| V(D)J 1 Primer Mix | 3.1 μL | 0.3 μM | |
| Total | 51.2 μL |
V(D)J amplification 1 reaction mix
- Mix gently and briefly spin down.
PCR cycling
Run the following program:
| Step | Temperature | Duration | Cycles | |
| Initial denaturation | 95 °C | 3 min | 1 | |
| Denaturation | 98 °C | 20 s | 12–15* | |
| Annealing | 62 °C | 30 s | ||
| Extension | 72 °C | 1 min | ||
| Final extension | 72 °C | 1 min | 1 |
PCR programm design
*Use 12 cycles as default; increase up to 15 cycles for low T cell content or low DNA yield.
V(D)J Amplification 1 Cleanup
Purify the first V(D)J amplification using AMPure XP beads.
- Vortex AMPure XP beads thoroughly.
- Add 46.1 μL AMPure XP beads (0.8X) to each PCR reaction.
- Mix by pipetting 15 times.
- Incubate 5 min at room temperature.
- Place tubes on the magnet (high) until the solution clears.
- Carefully remove and discard the supernatant.
- Add 200 μL 80% ethanol to the pellet and incubate ~30 s.
- Remove ethanol.
- Repeat the ethanol wash for a total of 2 washes.
- Air-dry the pellet for 2-3 min.
- Remove from the magnet and add 10 μL EB buffer.
- Resuspend thoroughly by pipetting ~15 times.
- Incubate 5 min at room temperature.
- Place on the magnet (low) until the solution clears.
- Transfer 10 μL to new tubes. This is the template for V(D)J Amplification 2.
V(D)J Amplification 2 (inner PCR)
This second nested PCR further enriches TCR V(D)J sequences and introduces inner primers.
a. Prepare V(D)J 2 primer mix on ice
| Primer | Volume | |
| 100 μM TRAC_3UTR_2 | 1 μL | |
| 100 μM HTCR_i_alpha | 1 μL | |
| 100 μM TRBC1_3UTR | 1 μL | |
| 100 μM TRBC2_3UTR | 1 μL | |
| 100 μM HTCR_i_beta | 1 μL | |
| H₂O | 5 μL | |
| Total | 10 μL |
V(D)J 2 primer mix
b. Prepare V(D)J Amplification 2 reaction mix on ice (25 μL reaction)
| Component | 25 μL reaction | Final conc. | |
| cDNA template (from step 10) | 10 μL | — | |
| 2X KAPA HiFi HotStart ReadyMix | 12.5 μL | 1X | |
| V(D)J 2 Primer Mix | 1.5 μL | 0.3 μM | |
| H₂O | 1 μL | N/A |
V(D)J 2 amplification mix
- Mix gently, spin down briefly.
PCR cycling
| Step | Temperature | Duration | Cycles | |
| Initial denaturation | 95 °C | 3 min | 1 | |
| Denaturation | 98 °C | 20 s | 10 | |
| Annealing | 62 °C | 30 s | ||
| Extension | 72 °C | 1 min | ||
| Final extension | 72 °C | 1 min | 1 |
PCR programm design
V(D)J Amplification 2 Cleanup
- Vortex AMPure XP beads thoroughly.
- Add 22.5 μL AMPure XP beads (0.9X) to each PCR reaction.
- Mix by pipetting 15 times.
- Incubate 5 min at room temperature.
- Place tubes on the magnet (high) until the solution clears.
- Remove and discard the supernatant.
- Add 200 μL 80% ethanol to the pellet; incubate ~30 s.
- Remove ethanol.
- Repeat the ethanol wash for a total of 2 washes.
- Air-dry the pellet for 2-3 min.
- Remove from magnet and add 20 μL EB buffer.
- Resuspend thoroughly (15× pipetting).
- Incubate 5 min at room temperature.
- Place on the magnet (low) until the solution clears.
- Transfer 20 μL to new tubes. This is your enriched V(D)J cDNA for library construction.
V(D)J Amplification QC
- Qubit dsDNA HS assay – quantify DNA.
- Agilent TapeStation HS D5000 – confirm size distribution and enrichment of V(D)J products.
Amplified TCR cDNA library from human PBMCs. Initial cDNA was generated with the 3’GEX v3.1 assay (10X Genomics)
V(D)J Library Construction
Proceed to “V(D)J Library Construction” from Chromium Next GEM Single Cell 5' v2 (Dual Index)
Input: 2-3 ng
Fragmentation, End Repair & A-tailing
a. Determine the volume for 25% concentration of sample. Dispense the sample volume in a tube on ice. If the volume required is less than 20 μL, adjust the total volume of each sample to 20 μL with nuclease-free water.
b. Vortex Fragmentation Buffer. Verify there is no precipitate.
c. Prepare Fragmentation Mix on ice. Pipette mix and centrifuge briefly.
| Component | PN | 1X (μL) | 4X + 10% (μL) | |
| Nuclease-free Water | - | 15 | 66 | |
| Fragmentation Buffer | 2000091 | 5 | 22 | |
| Fragmentation Enzyme | 2000090/2000104 | 10 | 44 | |
| Total | - | 30 | 132 |
Fragmentation Mix
d. Add 30 μl Fragmentation Mix into each tube containing 20 μl sample.
e. Pipette mix 15x (pipette set to 30 μl) on ice. Centrifuge briefly.
f. Incubate (Lid Temperature: 65 °C):
| Step | Temperature | Duration | |
| Fragmentation | 32 °C | 2 min | |
| End repair & A-tailing | 65 °C | 30 min | |
| Hold | 4 °C | hold |
Fragmentation thermal programm
Adaptor Ligation
a. Prepare adapter ligation mix
For each sample:
| Component | PN | 1X (μL) | 4X + 10% (μL) | |
| Ligation Buffer | 2000092 | 20 | 88 | |
| DNA Ligase | 220110/220131 | 10 | 44 | |
| Adapter Oligos | 2000094 | 20 | 88 | |
| Total | — | 50 | 220 |
Adaptor Ligation Mix
Mix well by pipetting and spin down briefly.
b. Ligation reaction
- Remove samples from the thermal cycler.
- Add 50 μL Adapter Ligation Mix to each 50 μL fragmented sample (total 100 μL).
- Mix by pipetting 15× (pipette set to 90 μL).
- Spin down briefly.
c. Incubation
Incubate in a thermal cycler (lid at 30 °C):
| Step | Temperature | Duration | |
| 1 | 20 °C | 15 min | |
| 2 | 4 °C | hold |
Adaptor Ligation programm
Post Adaptor Ligation Cleanup - SPRIselect
- Vortex SPRIselect reagent thoroughly.
- Add 0.8X μL SPRIselect (80 μL) to each 100 μL ligation reaction.
- Mix by pipetting 15× (pipette set to 150 μL).
- Incubate 5 min at room temperature.
- Place tubes on the magnet (high) until the solution is clear.
- Remove and discard the supernatant.
- Add 200 μL 80% ethanol to the pellet; wait 30 s.
- Remove ethanol.
- Repeat the ethanol wash for a total of 2 washes.
- Briefly centrifuge and return the tubes to the magnet (low).
- Remove any residual ethanol and air-dry the pellet for 2 min.
- Remove tubes from the magnet and add 30.5 μL EB buffer.
- Resuspend beads thoroughly by pipetting 15× (continue mixing if beads appear clumpy).
- Incubate 5 min at room temperature.
- Place on the magnet (low) until the solution clears.
- Transfer 30 μL of the eluate to a new tube strip. This is the adapter-ligated DNA for indexing PCR.
Sample Index PCR
This step adds sample indices and completes library amplification.
a. Choose sample indices
Select appropriate sample index primers such that no indices overlap between samples in a multiplexed run.
b. Prepare Sample Index PCR reaction mix (example for sample 1)
| Component | Volume | |
| Amp Mix (PN-2000047/2000103) | 50 μL | |
| P7_TRAC_3UTR_f3_i7_A1 (10 μM) | 3 μL | |
| P7_TRBC1_3UTR_f_i7_A1 (10 μM) | 3 μL | |
| P7_TRBC2_3UTR_f_i7_A1 (10 μM) | 3 μL | |
| P5_SI-TT-A1 (10 uM) | 3 μL | |
| H₂O | 8 μL | |
| Total | 70 μL |
Sample Index PCR mix
(For additional samples, use the corresponding index primer pairs A2, A3, A4, etc.)
c. PCR setup
- Add 70 μL Sample Index PCR Reaction Mix to 30 μL of adapter-ligated DNA (total 100 μL).
- Mix gently and spin down.
d. PCR cycling
Run the following program (lid at 105 °C):
| Step | Temperature | Duration | Cycles | |
| Initial denaturation | 98 °C | 45 s | 1 | |
| Denaturation | 98 °C | 20 s | 8 | |
| Annealing | 54 °C | 30 s | ||
| Extension | 72 °C | 20 s | ||
| Final extension | 72 °C | 1 min | 1 |
Sample Index PCR thermal programm
Post Sample Index PCR Cleanup – SPRIselect
- Vortex SPRIselect reagent thoroughly.
- Add 0.9X SPRIselect (90 μL) to each 100 μL PCR reaction.
- Mix by pipetting 15× (pipette set to 150 μL).
- Incubate 5 min at room temperature.
- Place on the magnet (high) until the solution clears.
- Remove the supernatant carefully.
- Add 200 μL 80% ethanol; wait 30 s.
- Remove ethanol.
- Repeat the ethanol wash for a total of 2 washes.
- Briefly centrifuge and place back on the magnet (low).
- Remove any residual ethanol and air-dry the pellet for 2 min.
- Remove from the magnet and add 25.5 μL EB buffer.
- Resuspend beads by pipetting 15×.
- Incubate 5 min at room temperature.
- Place on the magnet (low) until the solution clears.
- Transfer 25 μL of the eluate to a new tube strip.
- Store libraries at 4 °C for up to 72 h or at −20 °C for long-term storage.
Post Sample Index PCR QC
Perform final library QC:
- Qubit dsDNA HS assay – for accurate concentration.
- Agilent TapeStation HS D1000 – to verify the expected library size distribution and absence of major contaminants.
TCR short-read sequencing library
Library Quantification and Sequencing
qPCR quantification
- Quantify final circVDJ-seq libraries using a suitable qPCR-based library quantification kit (e.g. for Illumina platforms), following the manufacturer’s instructions.
- Normalize libraries to the desired loading concentration for NextSeq 550 Mid-Output runs.
Sequencing run setup
- Sequence libraries on an Illumina System with the following custom read configuration: 90–28–10–10 (note that in contrast to the standard Illumina workflow, the library indices are sequenced in the last two sequencing cycles).
- Use the following custom sequencing primers:
| Primer name | Read (cycles) | Sequence (5' → 3') | |
| spTCR_Read1 (90) | VDJ sequence (90) | GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| spTCR_Read2 (28) | Cell barcode + UMI (28) | CATACTTGCTCCTACACGACGCTCTTCCGATCT | |
| spTCR_Read3 (10) | i5 Index (10) | AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC | |
| spTCR_Read4 (10) | i7 Index (10) | CAAGCAGAAGACGGCATACGAGATCTGAGTCAGTAGC*G |
Custom Sequencing Primers
Protocol references