Apr 28, 2026
Version 3
FLInt 2.0 integration in C. elegans (version3) V.3
- Nawaphat Malaiwong1,
- Michael O'Donnell1
- 1Yale University
- Protocol_NM


Protocol Citation: Nawaphat Malaiwong, Michael O'Donnell 2026. FLInt 2.0 integration in C. elegans (version3). protocols.io https://dx.doi.org/10.17504/protocols.io.eq2lyqm1pvx9/v3Version created by Nawaphat Malaiwong
Manuscript citation:
Nawaphat Malaiwong, Porhathai Malaiwong, Chloe Kim and Michael O’Donnell, FLInt 2.0: Robust and customizable single shot integration in C. elegans, 2026, submitted
Nawaphat Malaiwong, Montserrat Porta-de-la-Riva, Michael Krieg, FLInt: single shot safe harbor transgene integration via Fluorescent Landmark Interference, G3 Genes|Genomes|Genetics, Volume 13, Issue 5, May 2023, jkad041, https://doi.org/10.1093/g3journal/jkad041
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: April 12, 2026
Last Modified: April 28, 2026
Protocol Integer ID: 314873
Keywords: C. elegans, transgene integration, array integration, CRISPR, gene editing, features of transgenic worm, transgenic worm, new version of flint, original flint method, flint, designated crispr, cas9 cutting site within the h2b region, chromosomal integration
Funders Acknowledgements:
NIH director's new innovator award
Grant ID: DP2-GM154014
Abstract
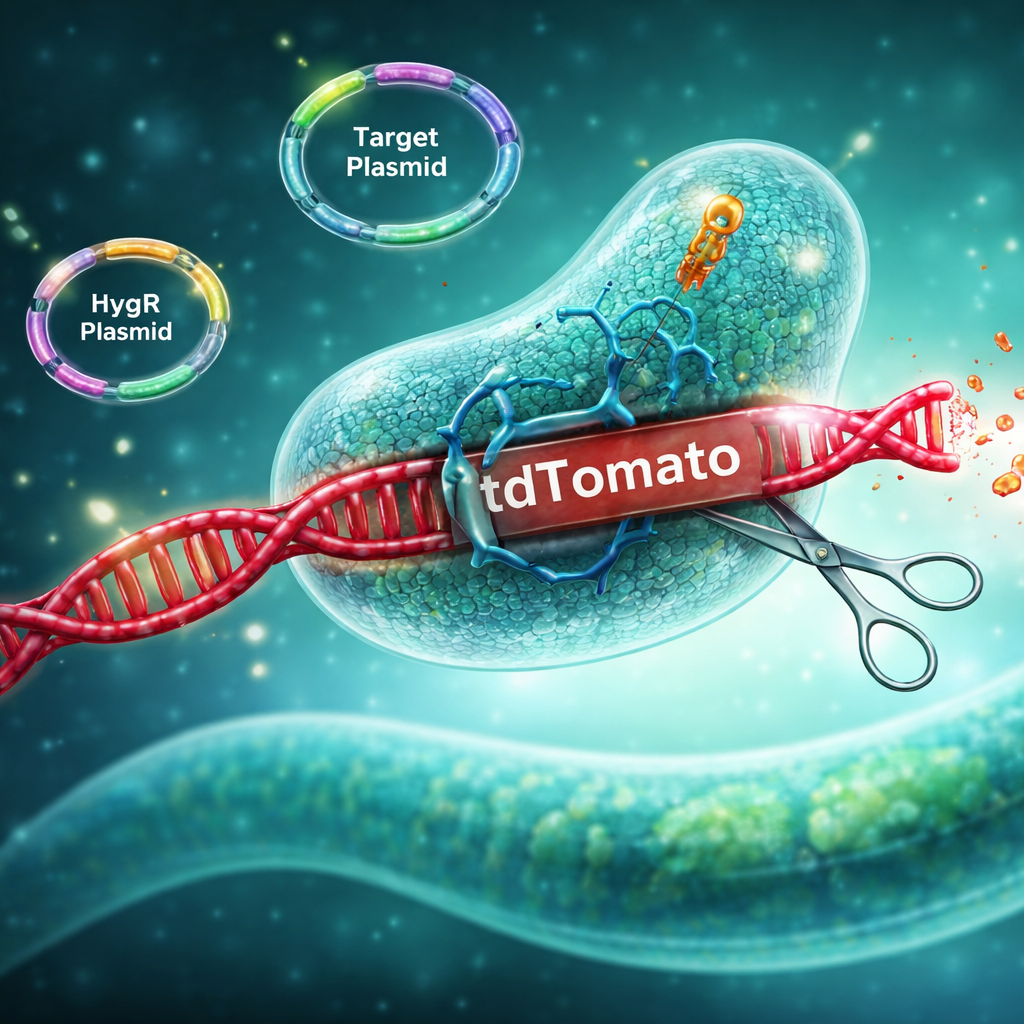
This protocol is adapted from the original FLInt method (Malaiwong et al., 2023) to enable chromosomal integration of multi-copy transgenic arrays in C. elegans. The updated version incorporates enhanced selection features that improve discrimination between true integrants and false positives (Malaiwong et al., 2026). The procedure reflects extensive troubleshooting and optimization performed during method development.
Materials
Worm strains for FLInt integration
| A | B | C | D | |
| Strains | landing locus | Genotypes | Available from | |
| EG7837 | Chr I | unc-119(ed3) III; oxTi712 [eft-3p::tdTomato::H2B::unc-54 3'UTR + Cbr-unc-119(+)] I | CGC | |
| EG7866 | Chr II | unc-119(ed3) III; oxTi564 [eft-3p::tdTomato::H2B::unc-54 3'UTR + Cbr-unc-119(+)] II | CGC | |
| EG7898 | Chr III | unc-119(ed3) III; oxTi619 [eft-3p::tdTomato::H2B::unc-54 3'UTR + Cbr-unc-119(+)] III | CGC | |
| EG7917 | Chr IV | unc-119(ed3) III; oxTi616 [eft-3p::tdTomato::H2B::unc-54 3'UTR + Cbr-unc-119(+)] IV | CGC | |
| EG7944 | Chr V | unc-119(ed3) III oxTi553 [eft-3p::tdTomato::H2B::unc-54 3'UTR + Cbr-unc-119(+)] V | CGC | |
| EG7826 | Chr X | unc-119(ed3) III oxTi308 [eft-3p::tdTomato::H2B::unc-54 3'UTR + Cbr-unc-119(+)] X | CGC |
Considerations before using this method
- Dependence on Pre-engineered Landing Sites: FLInt is a CRISPR-based integration method that relies on pre-engineered fluorescent landing sites (tdTomato insertions). As a result, array integration is restricted to genomic loci available in existing strains, including more than 200 tdTomato-marked strains distributed by the Caenorhabditis Genetics Center.
- Genomic Locus Effects on Integration Efficiency: Integration efficiency can vary depending on the genomic position of the tdTomato locus. In general, loci located near the chromosomal center tend to yield higher integration efficiency compared to those located on chromosome arms (Malaiwong et al., 2023).
- Quality of the starting extrachromosomal array: This method typically yields integration efficiencies of approximately 5–10% or lower. A high number of transgenic F1 animals increases the probability of recovering true integrants. Therefore, robust injection technique and generation of stable, well-transmitted arrays are critical for success.
- Array composition and complexity: Array composition can influence both integration efficiency and selection accuracy. Simpler arrays can be integrated without carrier DNA and may provide higher selection specificity. However, inclusion of carrier DNA (e.g., DNA ladder) to achieve a total concentration of ~100 ng/µL can promote formation of more complex arrays from multiple circular DNA. Although this may reduce selection stringency, it can improve representation of desired genotypes from multiple transgenes.
- Risk of False Positives and Screening Strategy: Not all non-fluorescent F2 animals represent true integrants. Loss or reduction of fluorescence may arise from factors other than intended integration. Therefore, secondary validation (F3 screening) is essential. The use of co-injection markers is optional but can facilitate identification of true integrants (100% array transmission).
- Genetic Background Considerations (unc-119 System): The tdTomato::Cbr-unc-119(+) strains are maintained in the unc-119(ed3) III background. This mutation may influence certain developmental or behavioral phenotypes. Outcrossing to remove the unc-119(ed3) allele may be required prior to downstream applications (strains available from CGC soon). In addition, inclusion of crRNA targeting Cbr-unc-119(+) can be considered to eliminate the selectable marker after integration.
- Influence of Experimental Conditions: Experimental conditions can substantially influence integration efficiency. Higher culture temperature (25 °C), the use of linearized plasmid DNA, and the inclusion of ssDNA oligonucleotides may improve recovery under certain conditions.
- Conceptual Flexibility of FLInt 2.0 Design: FLInt 2.0 is based on the concept of disrupting a fluorescent coding sequence–UTR module to generate a visual phenotype upon array integration. There is no single rigid implementation of this strategy; variations such as single or multiple CRISPR target sites, as well as inclusion of additional reagents, can be adapted depending on experimental design.
Preparation of reagents
- crRNA for cutting the H2B site (5'-ATGCTAACTAGTTTACTTGC-3')
- crRNA for cutting Cbr unc-119(+) site (5'-TTATGCATCATATGAGTAGT-3') (Optional for double cutting sites)
- crRNA for cutting AmpR site (5'-ATTAATAGACTGGATGGAGG-3') (Optional for linearizing plasmids in vivo)
- tracrRNA (IDT), Cas9 (IDT)
- ssODN(1) (5'-GGGAACCAAGGCCGTCACCAAGTACACTTCCAGCAAGTAAACTAGTTAGAGGCGGATAAAGTTGCAGGACCACTTCTGCGCTCG-3')
- ssODN(2) (5'-TCTAGCTTCCCGGCAACAATTAATAGACTGGATGGTAGCATTACCATATACTGGGTGATACATAGTTGAACGTAGGATAATAAAGTAAACTAGTTAGCATTCGTAGAATTCCAACTGA-3')
- hygR plasmid (Addgene #190933)
- hygromycin B (Thermo Fisher Scientific, cat. #10687010)
- materials and equipment for microinjection
FLInt 2.0 mix preparation
Prepare the FLInt 2.0 ribonucleoprotein (RNP) (10µL)
- 1µL of 100µM crRNA(FLInt2.0*) (IDT) >>> final concentration of crRNA = 10µM
- 0.5µL of 200µM tracrRNA (IDT) >>> final concentration of tracrRNA = 10µM
- 1µL of 62uM Cas9 endonuclease (IDT) >>> final concentration of Cas9 = 6.2µM
- 7.5µL of nuclease-free water
*crRNA combinations
For single cut >> 10µM crRNA(H2B) + 10µM tracrRNA + 6.2µM Cas9
For double cut >> 10µM crRNA(H2B)+ 10µM crRNA(unc-119) + 20µM tracrRNA + 6.2µM Cas9
For linearlized plasmid >> crRNAs + 10µM crRNA(ampR) + 20µM tracrRNA + 6.2µM Cas9
(total volume = 10µL)
NOTE: Different concentrations of crRNAs in combination have not been empirically tested and could be optimized by the user
- Incubate at 37ºC for 30 minutes
- Aliquot 2µL** of FLInt mix into PCR tube and store at -20ºC for the further use
**The volume of the FLInt mix can be higher than 2
Injection mix preparation
- Prepare the injection mix according to the standard protocol (Plasmid of interest: x ng/μL, Co-injection marker: y ng/μL, 1kb Plus DNA ladder (Invitrogen) as needed to bring the total DNA concentration to 100ng/μL)
- (Optional) Add 1 μL of 100μM ssODN(1) and 1 μL of 100μM ssODN(2)
- Add 2μL of the FLInt2.0 RNP mix
- Add nuclease-free water to bring the final volume to 10μL
- Centrifuge the mixture at 12,000rpm for 5–10 minutes before injection
NOTE: The volume of the FLInt mix can be higher than 2
NOTE: Adding ssODNs does improve selection specificity and "may" promote better integration efficiency
P0 worm preparation
- Culture tdTomato-expressing worms (see ‘Materials’) at 16°C or 20°C to obtain healthy young adults
- Select well-fed, young adult worms using a paintbrush under a dissecting microscope
- Transfer worms using halocarbon oil and mount the worms onto 2% agarose pad on coverslip
NOTE: For double cutting sites using crRNAs (H2B + unc-119), the tdTomato strain should be outcrossed with N2 to eliminate the unc-119(ed3) allele.
The outcrossed strains will be generated and deposited to CGC
Microinjection
- Inject the mix into single or both gonads of P0 worms
- After injection, transfer 10–15 P0 worms to a 5-cm NGM plate seeded with OP50, incubate the plates at 25°C
!!! Good injection technique is the most critical factor for obtaining integrants. At 1–2 days post-injection, the number of array-positive F1 progeny can be assessed to determine whether the batch should be re-injected.
Hygromycin selection
(Requires coinjection of hygR resistant plasmid)
Three days post-injection, apply 500μL of 5 mg/mL hygromycin B to each plate, allow the plates to dry on the benchtop for approximately 30 minutes (or less), then return them to the 25°C incubator
Selection of candidate worms
- On day 6 post-injection, identify healthy, non-tdTomato, co-injection marker-positive adult worms
- Transfer these candidate worms to individual small NGM/OP50 plates and culture them at 25°C
!!! Without hygromycin selection, worm chunking at F1 generation might be required to maintain the growing population
!!! Some worms might carry dim tdTomato expression. Please make sure you pick complete dark worms
!!! The succesful integration typically produces a large number of dark worms (10-40 candidates). Very few dark worms is indicative of a poor injection and unlikely to yield integrants.
Screen for integrated lines
On day 9 post-injection, identify plates with 100% co-injection marker expression in the progeny. These lines are potential integrants.
!!! Some plates may produce Unc worms due to aneuploidy (Yoshino et al.). This phenotype is typically associated with non-integrated animals or aberrant chromosomal arrangements. Such plates should be discarded.
Verification of transgene integration
Lines with uniform co-injection marker expression are considered candidate integrated lines. Confirm expression of the gene of interest via fluorescence or other relevant assays. If gene expression is not visually detectable, perform genotyping via PCR.
Designation of the integrated lines
Integrated lines derived from the same P0 plate are not considered independent due to the inability to trace individual lineages. However, separating injected P0 worms, or using several small pools of P0 animals after injection (described below), can enable the isolation of multiple distinct integrated lines.
Alternative procedures
- Hygromycin selection can be performed at any time after day 3 post-injection or during subsequent generations following the F1. Selection beyond day 3 is also feasible, as integrants can still remain within tdTomato-expressing populations. Researchers may choose an alternative time for hygromycin treatment based on the suitable time schedule. However, food supplementation might be required for animals recovering after drug selection.
- OP50 seeding can be adjusted based on the timing of screening and the expected number of worms. In the absence of hygromycin selection, the amount of OP50 on the plate should be carefully optimized. Since P0 worms produce both F1 and F2 progenies on the same plate—and homozygous integrants typically emerge in the F2 generation—it is essential to seed concentrated OP50 prior to the experiment to ensure a sufficient food source through F2 development. Additionally, 10-cm Petri dishes can be used to accommodate larger worm populations when necessary.
- Separating P0 worms onto individual plates after injection helps in accurately identifying integrated lines. Since multi-copy integrants on the same plate may originate from different P0 worms and thus represent distinct insertion sequences, tracking progeny may be crucial. Without such tracking, only one integrated line per plate should be registered to avoid confusion. Researchers should carefully consider whether obtaining multiple integrated lines from the same experiment is necessary based on their experimental goals.
- Culturing injected P0 worms at 25 °C is not essential, as incubation at 20 °C may promote better fitness in tdTomato-expressing strains. However, previous reports suggest that higher temperatures enhance integration efficiency (Frøkjær-Jensen et al., 2014).
- Based on Mello et al., adding single-stranded oligos might help integration efficiency. However, the role of oligos in integration process is still unclear.
Protocol references
Mello, Craig C., et al. "Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences." The EMBO journal 10.12 (1991): 3959-3970.
Frøkjær-Jensen, Christian, et al. "Random and targeted transgene insertion in Caenorhabditis elegans using a modified Mos1 transposon." Nature methods 11.5 (2014): 529-534.
Yoshina, Sawako, et al. "Locus-specific integration of extrachromosomal transgenes in C. elegans with the CRISPR/Cas9 system." Biochemistry and biophysics reports 5 (2016): 70-76.
Malaiwong, Nawaphat, Montserrat Porta-De-La-Riva, and Michael Krieg. "FLInt: single shot safe harbor transgene integration via F luorescent L andmark Int erference." G3: Genes, Genomes, Genetics 13.5 (2023): jkad041.
Malaiwong, Nawaphat, et al. "FLInt 2.0: Robust and customizable single shot integration in C. elegans." bioRxiv (2026): 2026-01.
Acknowledgements
The improvement of FLInt was conducted under the supervision of Mike O’Donnell at Yale University.