Dec 31, 2025
EZ-DMS - A Simple and Accessible Protocol and Software Package for Deep Mutational Scanning of Virus Proteins
- Gillman.Aaron.N 1,
- Samuel A McCarthy-Potter1,
- Rohith Rao Vujjini1,
- Madeline M Broghammer1,
- Cassian M Birler1,
- Alex B. Kleinpeter1,
- Hillel Haim1
- 1University of Iowa, Carver College of Medicine



Protocol Citation: Gillman.Aaron.N , Samuel A McCarthy-Potter, Rohith Rao Vujjini, Madeline M Broghammer, Cassian M Birler, Alex B. Kleinpeter, Hillel Haim 2025. EZ-DMS - A Simple and Accessible Protocol and Software Package for Deep Mutational Scanning of Virus Proteins. protocols.io https://dx.doi.org/10.17504/protocols.io.n2bvje19wgk5/v1
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: October 30, 2025
Last Modified: December 31, 2025
Protocol Integer ID: 231187
Keywords: Deep Mutational Scanning, Saturation Mutagenesis, Virus mutational landscape, Fitness Profiling, HIV-1, Envelope Glycoproteins, Antiviral Therapeutics, Temsavir, virus proteins mutation, software package for deep mutational scanning, deep mutational scanning, phenotypic potential of virus, viral genome replication, virus library, deep sequencing for genomic characterization, virus infectivity, site combinations in the same protein, virus, amino acid variant, frequency of each amino acid variant, bioinformatics, deep sequencing, genomic characterization, same protein, protein, phenotypic potential, amino acid preference, expertise in bioinformatics, amino acid change, infected culture, protein function, effects of all amino acid change
Funders Acknowledgements:
National Institutes of Health
Grant ID: R01 AI170205
Abstract
Mutations that occur during viral genome replication can result in new phenotypes, such as infection of new cell types or resistance to therapeutics. To determine the phenotypic potential of viruses, deep mutational scanning (DMS) can be used. This approach measures the effects of all amino acid changes at sites of interest on protein function, virus infectivity, or resistance to therapeutics. A virus library that contains all possible variants is produced and used to infect a culture of cells. The frequency of each amino acid variant in the infected culture, relative to the input stock, captures the relative fitness (preference) for each form under the selective pressure applied. Current DMS protocols are complex and costly, due to reliance on deep sequencing for genomic characterization and require expertise in bioinformatics to process and analyze the data. This limits the number of labs that can apply the technology. Here, we describe a simple and efficient protocol to perform DMS on single sites and on two-site combinations in the same protein. Dual-site DMS allows probing for epistatic and synergistic effects. Sequencing is performed using Oxford Nanopore technology, and the data are uploaded to a graphical user interface-based platform that calculates the amino acid preferences. The protocol does not require expertise in bioinformatics. This protocol can be completed in approximately 16 days, with 3-4 days of laboratory activity and 12-13 days of incubation, and at considerably lower costs than existing systems.
Guidelines
Given the broad utility of DMS to understand the evolutionary space of diverse viruses, we sought to increase the accessibility of this method to the broader scientific community. To this end, we developed a new step-by-step protocol with associated software to perform DMS on a single site or dual sites using replication competent HIV-1.
Materials
Biological Materials
Mix & Go! Competent Cells - DH5 Alpha ‱ (Zymo Research, T3007): E. coli DH5α Genotype: F-Φ80lacZΔM15 Δ(lacZYA-argF)U169 deoR nupG recA1 endA1 hsdR17(rK- mK+) phoA glnV44 (supE44) thi-1 gyrA96 relA1,λ-
Human Immunodeficiency Virus Type 1 (HIV-1): pNL4-3 AD8 (GenBank PV345784)
293T Cells (ATCC CRL-3216): Human Embryonic Kidney Cells:TZM-GFP Human Cell Line JC.53 Derived (BEI Resources, HRP-20041): HeLa Derived CD4+, CCR5+, CXCR4+, Tat/Rev-GFP
A3R5.7 Cells (BEI Resources, ARP-12386): Human T Lymphoblastic Leukemia, G418R CCR5 Expression
Bacterial
Luria Broth (RPI, L24400)
LB Agar (RPI, L24030)
Ampicillin (RPI, A40040)
Mix & Go! E. coli Transformation Kit (Zymo Research, T3001)
Cell Culture and Virus Production
DMEM High Glucose (Gibco, 11965092)
RPMI 1640 Medium (Gibco, 11875093)
FBS - Fetal Bovine Serum (Avantor, 89510-186)
Penicillin-Streptomycin 10,000 U/ml (Gibco, 15140122)
Trypsin-EDTA 0.25%, Phenol Red (Gibco, 25200056)
G418 Disulfate Solution 50 mg/ml (IBI Scientific, IB02060)
JetPrime (Polyplus, 101000046)
DNase-I (Roche, 4716728001)
1 M MgCl 0.22 μm sterile filtered (Sigma, M0250-500G)
CellPro‱ Vented Cell Culture Treated Flask (T75) 75 cm2 (Avantor, 10062-860)
6-Well Cell Culture Plates (Avantor, 10062-892)
96-Well Cell culture Plates (Corning, 3596)
0.45 μm Luer-Lock Polyethersulfone Syringe Filter (Avantor, 76479-020)
10 ml Luer-Lock Syringe (BD Diagnostic Systems, 302995)
Sucrose (RPI, S24065)
Phosphate-Buffered Saline (PBS) 10x pH 7.4 (Gibco, 70011069)
DNA and PCR Reagents
PrimSTAR Max 2x Master Mix (Takara, R045A)
QIAquick PCR Purification Kit (Qiagen, 28104)
Qiagen Miniprep Kit (Qiagen, 27104)
Zymoclean Gel DNA Recovery Kit (Zymo Research, D4001)
Zymo DNA Clean & Concentrator-5 (Zymo Research, D4013)
SmaI 10 U/μl (ThermoFisher, ER0661)
Tango Buffer (10X) (ThermoFisher, BY5)
AfeI (Eco47III) 10 U/μl (ThermoFisher, ER0322)
Buffer O (10X) (ThermoFisher, BO5)
rCutSmart‱ Buffer (New England Biolabs, B6004SVIAL)
In-Fusion‱ HD Cloning Kit (Takara, 639650)
OneTaq‱ 2X Master Mix with Standard Buffer (New England Biolabs, M0482S)
Molecular Grade Water (RPI, 248700)
Certified Molecular Biology Agarose (Bio-Rad, 1613101)
TAE Buffer 40 mM Tris-Acetate 1 mM EDTA (Ambion, AM9870)
NucleoBond Xtra Midi Plus (Macherey-Nagel, 740412.10)
SYBR‱ Safe DNA Gel Stain (Invitrogen, S33102)
BlueJuice‱ Gel Loading Buffer 10X (Invitrogen, 10816015)
RNA Extraction
Quick-RNA‱ Viral Kit (Zymo Research, R1034)
2-mercaptoethanol (Sigma, M6250)
Ethanol 100% (Decon Laboratories, 2716)
cDNA Synthesis
SuperScript IV Reverse Transcriptase (200 U/μl) (Invitrogen, 18090200)
5x SSIV Buffer (Invitrogen, 18090200)
0.1 M DTT (Invitrogen, 18090200)
10 mM dNTP mix (Invitrogen, 100011241)
Ribonuclease Inhibitor 40 U/μl (ThermoFisher, EO0381)
Molecular Grade Water (RPI, 248700)
Random Hexamers 50 ng/μl (Invitrogen 58875)
E. coli Ribonuclease H (RNase H) 2 U/μl (Invitrogen, 18021014)
Equipment
Thermal Cycler
Gel Electrophoresis System
Nanodrop Spectrophotometer
Microcentrifuge
Incubator
Ultracentrifuge (Optional)
Troubleshooting
Problem
General Cloning Issues
Solution
General cloning and transformation issues are best addressed through the troubleshooting provided by the manufacturer. However, in general ensuring purity of DNA, adjusting the ratio of DNA to cells, and ensuring plates are pre-warmed can improve transformation efficiency.
Problem
Inconsistent Sequencing Results
Solution
PCR amplification of Sequencing Amplicons needs to be optimized for each application. In general, sufficient cycles are needed to achieve adequate DNA, but over amplification can lead to bias and saturation. PCR should not proceed beyond the linear dynamic range of amplification. It may be necessary to take a small sample of the PCR every few cycles and quantitate by gel electrophoresis to determine optimal range. PCR can be performed in triplicates and pooled to further reduce amplification bias.
Problem
Low Sequencing Reads
Solution
Low counts returned from sequencing can be a result of cellular DNA contamination. To reduce contamination gel purification can be employed. Alternatively, PCR can be split into two phases with approximately 20 cycles using the cell lysate template followed by spiking a new PCR for an additional 8 cycles.
Safety warnings
HIV-1 is an infectious agent, comply with all BSL-2 plus additional containment practices mandated by law and good laboratory practices.
Ethics statement
All research involving HIV-1 mutagenesis must comply with applicable institutional, national, and international regulations governing biosafety and research.
Before start
This protocol describes an optimized and simple workflow to perform saturation mutagenesis for any HIV-1 protein. The principles described here are readily transferable to other viruses and infection models.
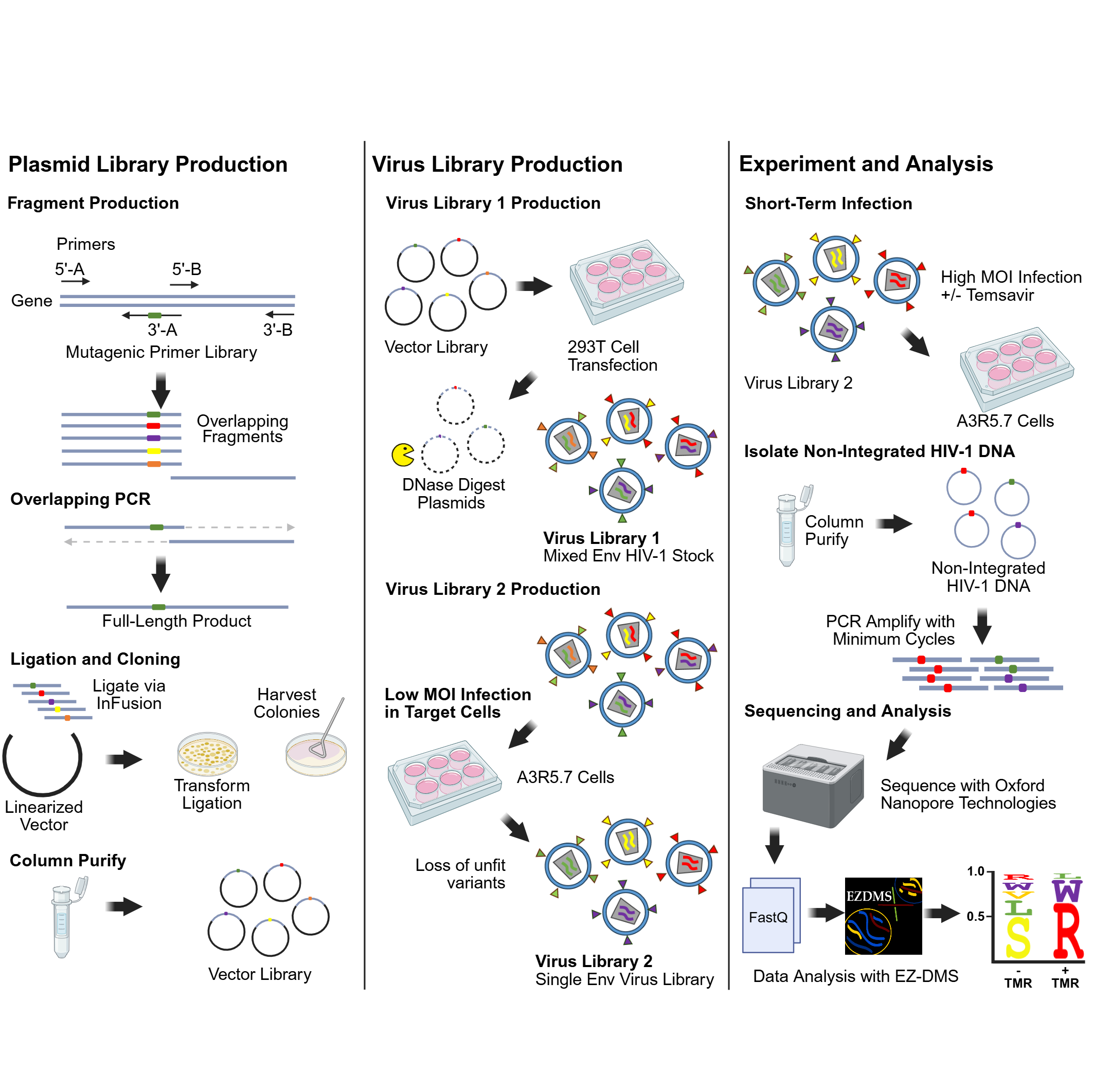
Plasmid Library Construction
Vector Linearization
Linearize the vector via PCR using a high-fidelity polymerase to remove 1000-3000 bp
around the site of mutagenesis and purify by gel purification. Primers are designed to include 15 bp overlaps with the insertion amplicon sequences. Primers include synonymous mutations to introduce blunt-end restriction enzyme cut sites to re-ligate the vector for storage (AfeI – 10 bp spacer – SmaI). Our primers and cycling conditions for pNLAD8 are detailed below. Dephosphorylation is not
required.
Note
It is recommended to construct a vector with a deletion of the region of interest that can be linearized by blunt-end digestion, particularly if you intend to introduce many mutations within that region. This approach can also mitigate potential PCR errors that arise from amplification of long repeat regions, such as the HIV-1 LTRs. Otherwise, linearization by PCR and gel purification is adequate.
Linearization Primers for HIV-1 Env
5’-ACTGCTGCCTAGCGCTTTTGTCATGAAACAAAC-3’
5’-AGGCAGCAGTATCCCGGGACCTAGAAAAACATG-3’
PCR Conditions
PCR Reaction Mixture
PrimeSTAR Max Premix 2x 25 μl
10 μM Forward Linearization Primer 1 μl
10 μM Reverse Linearization Primer 1 μl
Template (20 ng/μl) 1 μl
Molecular Grade Water 22 μl
PCR Cycling Conditions
98 °C 10 sec
TM-5 °C 10 sec
72 °C 10 sec/kb
24 Cycles
4 °C Hold
Perform a DpnI digest per the manufacturer's protocol to remove the template from the PCR product. Following the digestion, gel purify the linear vector using a gel purification kit per the manufacturer's instructions with elution in 30 μl Elution Buffer. Quantitate DNA via nanodrop.
Ligate the linear vector by In-Fusion and transform into Zymo Mix & Go! Competent Cells - DH5 Alpha™ per manufacturer’s protocol and incubate at 37 °C until colonies are visible.
In-Fusion Cloning Mix
pNLAD8ΔEnv Linearized (200 ng Total) X μl
5x In-Fusion Snap Assembly Master Mix 2.0 μl
Molecular Grade Water X μl
Total Volume 10 μl
Note
We utilized In-Fusion Cloning for our study, but any similar cloning protocol, such as NEB HiFi, should be readily substitutable.
Confirm the vector plasmid sequence and prepare an adequate quantity as needed via mini- or midi-plasmid preparation kits. At this stage, the vector can be linearized by restriction digest utilizing AfeI and SmaI per the manufacturer's protocol, column purified, and stored at ≤4 °C. Dephosphorylation is not required.
Amplicon Production
Amplify overlapping fragments containing your NNK site(s). Each overlap should be >20 bp. If introducing 1 NNK site, 1-2 fragments are needed, while 2-3 fragments are required for 2 NNK sites, depending on their sequence position. For reference, our fragments were constructed with the following primers and PCR conditions.
Note
Primers that introduce the NNK site should be long, up to 60 bp, with the NNK site located centrally with a long overlapping region (>20 bp). Synonymous mutations can be introduced around the NNK site for multiplexing.
Fragment Primers for HIV-1 EnvAD8-S375X, HIV-1 EnvAD8-M426X, and HIV-1 EnvAD8-S375X+M426X
Single Mutant: pNL-EnvAD8(375X) Primers
Upstream Fragment (1429 bp)
5’-TTTCATGACAAAAGCCTTAGGCATCTCC-3’
5’-GAATTACAGTAGAAAAATTCGCCACCACAATTAAAMNNGTGCATTACAATTTCTGGGTCC-3’
Downstream Fragment (1567 bp)
5’-GTGGTGGCGAATTTTTCTACTGTAATTCAACAC-3’
5’-GTTTTTCTAGGTCCCGAGATACTGCTCC-3’
Single Mutant: pNL-EnvAD8(426X) Primers
Upstream Fragment (1547 bp)
5’-TTTCATGACAAAAGCCTTAGGCATCTCC-3’
5’-GTTTATAATTTGTTTAATTCTACAGGGGAGTGTGATAGTGTC-3’
Downstream Fragment (1459)
5’-GTTTTTCTAGGTCCCGAGATACTGCTCC-3’
5’-CTATCACACTCCCCTGTAGAATTAAACAAATTATAAACNNKTGGCAAGAAGTAGGAAAAG-3’
Double Mutant: pNL-EnvAD8(375X+426X) Primers
Upstream Fragment (1429 bp)
5’-TTTCATGACAAAAGCCTTAGGCATCTCC-3’
5’-GAATTACAGTAGAAAAATTCGCCACCACAATTAAAMNNGTGCATTACAATTTCTGGGTCC-3’
Middle Fragment (146 bp)
5’-GTGGTGGCGAATTTTTCTACTGTAATTCAACAC-3’
5’-GTTTATAATTTGTTTAATTCTACAGGGGAGTGTGATAGTGTC-3’
Downstream Fragment (1459)
5’-CTATCACACTCCCCTGTAGAATTAAACAAATTATAAACNNKTGGCAAGAAGTAGGAAAAG-3’
5’-GTTTTTCTAGGTCCCGAGATACTGCTCC-3’
PCR Conditions
PCR Reaction Mixture
PrimeSTAR Max Premix 2x 12.5 μl
10 μM Forward Primer 1.5 μl
10 μM Reverse Primer 1.5 μl
Template (20 ng/μl) 0.5 μl
Molecular Grade Water 9 μl
Total volume 25 μl
PCR Cycling Conditions
98 °C 10 sec
98 °C 10 sec
TM-5 °C 15 sec
72 °C 10 sec/kb
25 Cycles
4 °C Hold
Gel purify the fragments using a gel purification kit per the manufacturer's instructions with elution in 30 μl Elution Buffer. Quantitate DNA via nanodrop.
Overlapping PCR
Combine the fragments at a 1:1 molar ratio (if of similar size) for a total of 300 ng of DNA. If one fragment is substantially smaller (<5x), combine at a 2:1 molar ratio.
When combining two fragments for a single NNK site, set up the reaction detailed below:
PCR Reaction Mixture
PrimeSTAR Max Premix 2x 12.5 μl
Fragment 1 X μl
Fragment 2 X μl
Molecular Grade Water to 25 μl total volume
Total volume 25 μl
PCR Cycling Conditions
98 °C 30 sec
98 °C 15 sec
TM* °C 15 sec
72 °C 10 sec/kb
10 Cycles
98 °C 15 sec
TM-5* °C 15 sec
72 °C 10 sec/kb
10 cycles
4 °C Hold
*Use the overlapping region between the fragments to determine the TM.
When combining three fragments for two NNK sites, set up the reaction detailed below:
PCR Reaction Mixture
PrimeSTAR Max Premix 2x 12.5 μl
Fragment 1 X μl
Fragment 2 X μl
Fragment 3 X μl
Molecular Grade Water to 25 μl total volume
Total volume 25 μl
PCR Cycling Conditions
98 °C 30 sec
98 °C 15 sec
TM* °C 15 sec
72 °C 10 sec/kb
10 Cycles
98 °C 15 sec
TM-5* °C 15 sec
72 °C 10 sec/kb
10 cycles
4 °C Hold
*Use the overlapping region between the fragments to determine the TM.
Following the overlapping PCR reactions, column purify using PCR or DNA purification kit per manufacturer’s protocol with resuspension in 30 μl of Elution Buffer.
Amplicon Amplification
Perform another round of PCR using the overlapping PCR product as a template. Use the amplicon flanking primers to amplify the full-length amplicon. Prepare a 75 μl master mix and split it between three 25 μl PCR reactions.
Amplicon flanking primers
5’-TTTCATGACAAAAGCCTTAGGCATCTCC-3’
5’-GTTTTTCTAGGTCCCGAGATACTGCTCC-3’
PCR Conditions
PCR Reaction Mixture
PrimeSTAR Max Premix 2x 37.5 μl
10 μM Forward Primer 4.5 μl
10 μM Reverse Primer 4.5 μl
Template 15 μl
Molecular Grade Water 13.5 μl
Total volume 75 μl
PCR Cycling Conditions
98 °C 30 sec
98 °C 15 sec
TM °C 15 sec
72 °C 10 sec/kb
4 Cycles
98 °C 15 sec
TM-5 °C 15 sec
72 °C 10 sec/kb
20 cycles
4 °C Hold
Following the PCR reaction, pool the samples and purify using a PCR purification kit per the manufacturer's protocol, with elution in 30 μl elution buffer. Quantitate DNA via Nanodrop.
Plasmid Library Assembly
Setup the In-Fusion reaction with 200 ng of total DNA at a 1:2 molar ratio of linearized vector to library amplicon. Include a negative control with no insertion product. Incubate for 15 min at 50 °C, then place on ice.
In-Fusion Cloning Mix
Vector Linearized X μl
Library Amplicon X μl
5x In-Fusion Snap Assembly Master Mix 2.0 μl
Molecular Grade Water to 10 μl total volume
Thaw 100 μl Zymo Mix & Go! Cells on ice for 5-10 minutes, add 5 μl of the In-Fusion reaction and incubate for 2-5 minutes before plating directly onto 1 to 2 pre-warmed LB agar plates with antibiotics. Adjust volumes and plate size for the appropriate number of colonies needed.
Note
Protein synthesis inhibiting antibiotics, such as tetracycline or kanamycin, require 1 hour recovery in antibiotic free SOC media before plating.
Count the colonies to ensure that there are sufficient colonies for complete coverage. The 95% joint probability for selecting enough colonies to have every amino acid present, accounting for synonymous NNK codons, is 172 colonies for single mutants and 8,128 colonies for double mutants.
(Optional) Confirm ligation efficacy by colony PCR using amplicon flanking primers.
Add 5 ml pre-warmed LB broth with antibiotics to the top of each plate and scrape colonies.
(Optional) Increase volume of harvested colonies to 50 ml and recover cells for 1-2 hours at 37 °C with shaking to increase yield. Avoid growing cells >2 hours as it may bias plasmid distribution.
Pellet the cells and freeze the pellet at -20 °C, or purify the plasmid library using mini- or midi-prep kits per manufacturer’s protocol, depending on size of pellet.
To confirm plasmid library diversity, PCR amplify an approximately 1000 bp region containing the NNK site(s) for sequencing via Oxford Nanopore Technologies. Our PCR reaction for generating amplicons for sequencing is detailed below. Perform PCR in triplicate and pool for double NNK mutant libraries.
Sequencing Primers for HIV-1 Env S375X and M426X
5’-TGTCAACTCAACTGCTGTTAAATGG-3’
5’- CCTCAGCAAGTTGTTCTGCTG -3’
Sequencing PCR Reaction
PrimeSTAR Max Premix 2x 12.5 μl
10 μM Forward Primer 1.5 μl
10 μM Reverse Primer 1.5 μl
Plasmid library (20 ng/μl) 1 μl
Water 8.5 μl
Total volume 25 μl
Sequencing PCR Cycling Conditions
98 °C 10 sec
56.9 °C 10 sec
72 °C 10 sec/kb
24 Cycles
4 °C Hold
Virus Library 1 Production: Transfection of the Plasmid Library
Seed 6-well cell culture plates with 5x105 293T cells per well in 2 ml Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS) and 100 U/ml penicillin-streptomycin (Complete DMEM). Incubate the plates at 37 °C with 5% CO2 for 14-18 hours until cells are 50% confluent.
Transfect 293T cells with 2 μg of the plasmid library per well with JetPrime, or a similar transfection reagent, per manufacturer’s protocol.
Note
Changing the media 12-16 h post transfection improves cell viability.
Harvest supernatant between 40-48 hours post-transfection. Centrifuge at 800 x g for 5 minutes to pellet cell debris and filter the supernatant through a 0.45 μm filter syringe filter.
Add DNase-1 to a final concentration of 100 U/ml and MgCl2 to 10 mM to the filtered supernatant and incubate for 30 minutes at 37 °C.
Aliquot Virus Library 1 (VL1) stocks and flash freeze in a dry-ice ethanol bath. Store aliquots at -80 °C.
Quantify the virus via TCID50 assay utilizing TZM-GFP positive cells in Complete DMEM supplemented with 20 μg/ml DEAE-Dextran. Seed 96-well plates with 2.8x104 TZM-GFP cells per well in 175 μl Complete DMEM 16-18 hours prior to infection to achieve approximately 70% initial confluency. Allow the infection to proceed for 3 days, then quantify the plate via fluorescent microscopy.
Virus Library 2 Production: Passaging VL1
For HIV-1, the passage of the Virus Library 1 to produce Virus Library 2 (VL2) should be done in the cell type used in subsequent assays as there will be selection bias against unfit variants. For our applications, we used A3R5.7 Human T-Lymphoblastic Leukemia cells expressing CCR5, but other HIV-1 infectable cells should be readily substitutable.
Note
HIV-1 contains two genome copies per virion, thus the virions produced by transfection will be heterogenous for NNK sites. A low MOI passage of the initial transfection produced virus is required to obtain a library of virus with a single genotype per virion. This step is not necessary for viruses that contain a single copy of their genome.
Infect 2x106 A3R5.7 cells in a 6-well plate with 2 ml total volume of RPMI 1640 Medium containing 10% FBS, 100 U/ml penicillin-streptomycin, 1 mg/ml G418 disulfate (Complete RPMI-G418), and 20 μg/ml DEAE-Dextran with an MOI of 0.003 TZM-GFP IU/ml for single NNK mutants or an MOI of 0.006 TZM-GFP IU/ml for double NNK mutants. Incubate for 24 h at 37 °C and 5% CO2.
24 hours post-infection, pellet the cells at 800 x g for 5 mins and replace the media with Complete RPMI-G418 and 20 μg/ml DEAE-Dextran.
At 72 h post-infection, centrifuge cells at 800 x g for 5 mins to pellet cell debris and filter the supernatant through a 0.45 μm filter syringe filter.
(Optional) Concentrate the virus by ultra-centrifugation over a 30% sucrose gradient at 150,000 x g for 1 hour at 10 °C under vacuum. Concentrate virus >20 fold by resuspending in PBS, allow the virus 30 minutes with gentle intermittent mixing at room temperature to completely resuspend.
Aliquot virus stocks and flash freeze in a dry-ice ethanol bath. Store aliquots at -80 °C.
Quantify the virus via TCID50 assay utilizing TZM-GFP positive cells in Complete DMEM supplemented with 20 μg/ml DEAE-Dextran. Seed 96-well plates with 2.8x104 TZM-GFP cells per well in 175 μl Complete DMEM 16-18 hours prior to infection to achieve approximately 70% initial confluency. Allow the infection to proceed for 2-3 days, then quantify the plate via fluorescent microscopy.
Sequencing Input Library
Use one aliquot of the Virus Library 2 stock for sequencing the input library. Purify the viral RNA via column purification per the manufacturer’s protocol. Elute the purified RNA in the maximum volume so that the entire sample can be used as a template for cDNA synthesis. In this protocol, we used Zymo Quick RNA-Viral kit (Zymo Research) with elution in 11 μl molecular grade water.
Synthesize cDNA from the purified viral RNA using a reverse transcription kit. We used SuperScript IV (Invitrogen) per the manufacturer’s protocol with Random Hexamers (50 ng/μl). Use the entire purified RNA sample for this step. Our first strand synthesis reaction is outlined below.
Denature Template Reaction
Random Hexamers (50 ng/μl) 1 μl
10 mM dNTP Mix 1 μl
RNA Template (Elution) 11 μl
Total Volume 13 μl
Briefly centrifuge and run in thermocycler under the following conditions.
Thermocycler Conditions
65 °C 5 min
4 °C Hold
Prepare the Buffer Reaction as detailed below.
Buffer Reaction
Denatured Template Reaction 13 μl
Ribonuclease Inhibitor (40 U/μl) 1 μl
100 mM DTT 1 μl
5x SuperScript IV Buffer 4 μl
SuperScript IV Reverse Transcriptase (200 U/μl) 1 μl
Total Volume 20 μl
Briefly centrifuge and run in thermocycler under the following conditions.
Cycling Conditions
23 °C 10 min
50 °C 10 min
80 °C 10 min
4 °C Hold
Perform sequencing PCR as described below to generate amplicons for sequencing. Use the entire cDNA sample as the template for PCR amplification. If sequencing a double mutant library, perform the PCR reaction in triplicate and pool afterwards to minimize amplification bias.
Non-Index Sequencing Primers for HIV-1 Env S375X and M426X
5’-TGTCAACTCAACTGCTGTTAAATGG-3’
5’- CCTCAGCAAGTTGTTCTGCTG -3’
Sequencing PCR Reaction
PrimeSTAR Max Premix 2x 12.5 μl
10 μM Forward Primer 1.5 μl
10 μM Reverse Primer 1.5 μl
Template (20 ng/μl) 1 μl
Water 8.5 μl
Total volume 25 μl
Sequencing PCR Cycling Conditions
98 °C 30 sec
TM-5 °C 10 sec
72 °C 10 sec/kb
24 Cycles
4 °C Hold
Following PCR, column purify using PCR or DNA purification kit per manufacturer’s protocol with resuspension in 30 μl of Elution Buffer.
Experimental Application of Library
Given the critical role of the AD8-Env 375 and 426 sites in CD4 receptor binding and temsavir resistance, we evaluated the impact of a stringent bottleneck on initial viral entry using the IC99 of temsavir (250 nM) as determined in AD8 pseudovirus. Herein, we describe our protocol and evaluation as an example. However, evaluation of integration, viral production, or viral passage can be readily evaluated if the genome can be PCR amplified.
For each sample, prepare a 1 ml infection mixture containing Complete RPMI-G418 supplemented with 40 μg/ml DEAE-Dextran with an MOI of 0.01-0.1 TZM-GFP IU/ml of HIV-1 AD8-Env375X or AD8-Env375X426X viral library in the presence or absence of 250 nM temsavir. Prior to infection, pre-incubate viral libraries in the presence of temsavir for 30 minutes at 37 °C.
Pellet 1x106 A3R5.7 cells per sample and resuspend in infection mixture and incubate in a 6-well plate for 3 hours at 37 °C with 5% CO2.
3 h post infection, pellet cells at 800 x g for 5 minutes and resuspend in fresh Complete RPMI-G418 without DEAE-Dextran and incubate for an additional 13-15 hours in a 6-well plate at 37 °C with 5% CO2.
At 16-18 hours post-infection, pellet cells at 800 x g for 5 minutes, and purify non-integrated DNA from the cells using Qiagen Miniprep kit following manufacturer’s protocol, substituting the bacterial pellet for the A3R5.7 cell pellet from one well per column with elution in 100 μl of 55 °C Elution Buffer.
PCR amplify a ~1000 bp region surrounding the NNK site by Nested PCR as described below to generate amplicons for sequencing. We included indexing primers that vary by 1 bp at the 5’ end for multiplexing of the data.
Note
Samples are multiplexed in groups of three per sequencing run to obtain at least 1000 reads per sample for amino acid frequency estimation. Double mutants are sequenced individually to achieve at least 3000 reads per sample. Multiplexing depends on total read yield and sequencing type to ensure adequate read depth.
Outer Nested Primers for Env
5’-GGCATCTCCTATGGCAGGAAGAAG-3’
5’-GTCTCGAGATACTGCTCCCACCC-3’
Nested-PCR Reaction Mixture per Amplicon
PrimeSTAR Max Premix 2x 12.5 μl
10 μM Forward Primer 1 μl
10 μM Reverse Primer 1 μl
Template (Elution) 10.5 μl
Total volume 25 μl
Sequencing PCR Cycling Conditions
98 °C 30 sec
59.8 °C 10 sec
72 °C 10 sec/kb
18 Cycles
4 °C Hold
Following PCR, column purify using a PCR or DNA purification kit per manufacturer’s protocol with resuspension in 30 μl of Elution Buffer.
Perform second round of PCR as described below, utilizing one Forward Index Primer with the Reverse Universal Primer for each sample.
Note
Instead of the standard TM-5 °C, we employed TM-2 °C for increased specificity.
Index Sequencing Primers for HIV-1 Env S375X and M426X
Forward Index Primers for Env
5’- CCAGTACTGTCAACTCAACTGCTGTTAAATGG-3’
5’- CCAGTATTGTCAACTCAACTGCTGTTAAATGG-3’
5’- CCAGTAATGTCAACTCAACTGCTGTTAAATGG-3’
N – Denotes unique sequence.
Reverse Universal Primer for Env
5’- CCTCAGCAAGTTGTTCTGCTG -3’
Nested-PCR Reaction Mixture per Amplicon
PrimeSTAR Max Premix 2x 12.5 μl
10 μM Forward Index Primer 1 μl
10 μM Reverse Universal Primer 1 μl
Nested-PCR Elution 10.5 μl
Total volume 25 μl
Sequencing PCR Cycling Conditions
98 °C 30 sec
59.9 °C 10 sec
72 °C 10 sec/kb
18 Cycles*
4 °C Hold
*Cycles may need to be increased, use minimal cycles as possible to reduce bias.
Gel purify fragments by gel electrophoresis using a gel purification kit per manufacturer’s protocol with elution in 20 μl Elution Buffer. Quantitate DNA via Nanodrop.
Sequence non-integrated DNA amplicons and upload the FastQ files to the EZ-DMS software to determine amino acid frequency and preferences of the output virus.
EZ DMS Software: Determining Preferences
Download EZ-DMS Software available from the following web address.
Note
EZ-DMS Software program calculates the enrichment ratio (𝜙) for each amino acid 𝑎 at position 𝑝:
𝜙𝑝,𝑎=(ƒ𝑝,𝑎(𝑐𝑒𝑙𝑙)/ƒ𝑝,𝑤𝑡(𝑝)(𝑐𝑒𝑙𝑙))/(ƒ𝑝,𝑎(virus)/ƒ𝑝,𝑤𝑡(𝑝)(virus))
where ƒ𝑝,𝑎(𝑐𝑒𝑙𝑙) and ƒ𝑝,𝑤𝑡(𝑝)(𝑐𝑒𝑙𝑙) are the frequencies in the cell lysate of amino acid 𝑎 or the wild-type (𝑤𝑡) amino acid for position 𝑝, and ƒ𝑝,𝑎(virus) and ƒ𝑝,𝑤𝑡(𝑝)(virus) are the frequencies of these forms in the input virus sample used for infection.
Finally, we calculated the amino acid preference (𝜋) for each
where the ∑𝑎′𝜙𝑝,𝑎′ is the sum of enrichments for all amino acids 𝑎′ at position 𝑝.
Launch software and proceed to Preferences option.
Upload Reference sequence in .fasta format with variable sites listed as NNK.
Upload .fastQ file of input virus (VL2) sequencing to Data sequence 1.
Upload .fastQ file for output virus sequencing to Data sequence 2.
Name output file for your reference.
Select the number of nucleotides flanking either side of the NNK that must match with the phred score threshold to filter unreliable sequences.
Select number variable sites in your amplicon.
Select the wildtype amino acid for each NNK site.
If multiplexing, enter a barcode. Multiple barcodes can be used for each Data sequence file.
Submit the files to generate a comma-separated values (CSV) file containing the amino acid and corresponding preferences based on the quality thresholds.
(Optional)The sum of the preferences equals 1. To normalize preferences so that a preference of 1 is no change, multiply the values by 21.
EZ DMS Software: Determining Site Count and Frequency
Launch software and proceed to Variable Site option.
Upload Reference sequence in .fasta format with the variable site(s) listed as NNK.
Upload .fastQ file from sequencing.
Name output file for your reference.
Select the number of nucleotides flanking either side of the NNK site that must match with the phred score threshold to filter unreliable sequences.
Select the number of variable sites in your amplicon.
Enter a barcode if multiplexing.
Submit the files to generate a comma-separated values (CSV) file will be generated containing the amino acid and corresponding counts based on the quality thresholds.