Oct 09, 2025
Version 2
Draft genome of the switchgrass head smut pathogen Tilletia maclaganii V.2
- Gian Maria Niccolò Benucci1,2,
- Kevin L. Myers3,
- Chathu Wijewardana1,
- Gary C. Bergstrom3,
- Stephen Mondo4,
- Robert Riley4,
- Anna Lipzen4,
- Jonathan Del Rosario4,
- Mi Yan4,
- Vivian Ng4,
- Igor V. Grigoriev4,5,
- Acer VanWallendael2,6,7,8,9,
- David B. Lowry2,6,7
- 1Department of Plant, Soil and Microbial Sciences, Michigan State University, East Lansing MI, 48824 USA;
- 2Great Lakes Bioenergy Research Center (GLBRC), 48824 East Lansing, MI USA;
- 3School of Integrative Plant Science, Plant Pathology and Plant-Microbe Biology Section, Cornell University, Ithaca, NY 14853-5904, USA;
- 4U.S. Department of Energy Joint Genome Institute, Lawrence Berkeley National Laboratory, Berkeley, CA, 94720, USA;
- 5Department of Plant and Microbial Biology, University of California Berkeley, Berkeley, CA, 94720, USA;
- 6Department of Plant Biology, Michigan State University, East Lansing MI, 48824 USA;
- 7Plant Resilience Institute, Michigan State University, East Lansing MI, 48824 USA;
- 8Department of Horticultural Science, North Carolina State University, Raleigh, NC, 27695;
- 9Department of Crop and Soil Science, North Carolina State University, Raleigh, NC, 27695



External link: https://doi.org/10.1128/mra.00991-25
Protocol Citation: Gian Maria Niccolò Benucci, Kevin L. Myers, Chathu Wijewardana, Gary C. Bergstrom, Stephen Mondo, Robert Riley, Anna Lipzen, Jonathan Del Rosario, Mi Yan, Vivian Ng, Igor V. Grigoriev, Acer VanWallendael, David B. Lowry 2025. Draft genome of the switchgrass head smut pathogen Tilletia maclaganii. protocols.io https://dx.doi.org/10.17504/protocols.io.dm6gpmr55gzp/v2Version created by gian benucci
Manuscript citation:
Benucci GMN, Myers KL, Wijewardana C, Bergstrom GC, Mondo S, Riley R, Lipzen A, Rosario JD, Yan M, Ng V, Grigoriev IV, VanWallendael A, Lowry DB Draft genome of the switchgrass head smut pathogen Tilletia maclaganii. Microbiology Resource Announcements 14(12). doi: 10.1128/mra.00991-25
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: October 09, 2025
Last Modified: October 09, 2025
Protocol Integer ID: 229436
Keywords: Smut, pathogen, genome, bioenergy, switchgrass, switchgrass head smut pathogen tilletia, significant pathogen of the bioenergy crop switchgrass, annotation of the first reference genome, first reference genome, draft genome, bioenergy crop switchgrass, significant pathogen, tilletia maclaganii, sequencing
Funders Acknowledgements:
Department of Energy
Grant ID: DE-SC0018409
Abstract
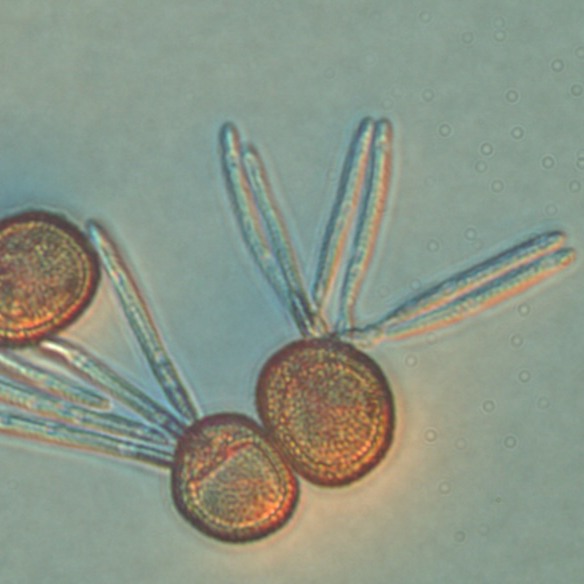
The head smut (Tilletia maclaganii) is a significant pathogen of the bioenergy crop switchgrass. T. maclaganii typically is more prevalent in older stands of switchgrass and can contribute to significant biomass loss. Here, we outline the methods for the sequencing, assembly, and annotation of the first reference genome for Tilletia maclaganii.
Image Attribution
Photo by Christine Layton
Guidelines
NA
Materials
DNA and RNA isolation:
- Macherey-Nagel Nucleobond HMW DNA kit
- New England Biolabs Monarch Total RNA Miniprep Kit (New England Biolabs, MA, USA)
- Thermo Scientific Nanodrop (Thermo Scientific, MA, USA)
- Agilent 5200 Fragment Analyzer (Agilent Technologies, Santa Clara, CA, USA)
Genome sequencing:
- PacBio Sequel IIe sequencing platform (Pacific Biosciences, CA, USA)
- Megaruptor 3 (Diagenode, Seraing, Belgium)
- SMRTbell Barcoded Adapter Plate 3.0 (Pacific Biosciences, CA, USA)
- SMRTbell Express Template Prep Kit 2.0 (Pacific Biosciences, CA, USA)
- AMPure PB Beads (Pacific Biosciences, CA, USA)
- BluePippin (Sage Science, MA, USA)
- SMRT Link 10.2, 8M v1 SMRT cells, Version 2.0 sequencing chemistry
Transcriptome sequencing:
- PerkinElmer Sciclone NGS robotic liquid handling system
- Illumina TruSeq Stranded mRNA HT sample prep kit
- KAPA Biosystems next-generation sequencing library qPCR kit (KAPA Biosystem, MA)
- Roche LightCycler 480 real-time PCR instrument (Roche, MA, USA)
- Illumina NovaSeq 6000 sequencing platform
- NovaSeq XP v1.5 reagent kits, S4 flow cell
Bioinformatic analysis:
- bbmap suite (http://sourceforge.net/projects/bbmap/)
- BBtools version 38.79 [kmercountexact.sh default; bbnorm.sh pigz passes=1 bits=16 target=9999999 min=174; bbduk.sh maxgc=0.4].
- Flye version 2.9-b1768 [-g 100k --asm-coverage 100 --pacbio-hifi]
- Prodigal verson 2.6.3 [-p meta]
- HMMER version 3.1b2 [--domtblout]
- BBDuk [bbduk.sh k=25 mm=f kmask=N]
- BBTools [k=25 mm=f mkf=0.03 ordered ow]
- RACON version 1.4.13 [-u -t 36]
- Trinity version 2.11.0
Safety warnings
NA
Ethics statement
NA
Before start
NA
Fungal isolation and DNA extraction
Tilletia maclaganii was isolated from switchgrass cultivar Shawnee in Ithaca NY, USA (42.4654130 N, -76.4590049 W) in 2021.
Approximately 100 mg of teliospores collected from infected switchgrass panicles were transferred to a 1.5 ml microcentrifuge tube. The teliospores were then surface sterilized by the addition of 1.0 ml of 0.165% (v/v) sodium hypochlorite, vortexing, and incubating for approximately 20 seconds. The teliospore suspension was then centrifuged for 10 seconds at 1000 X g, followed by removal of the supernatant with a pipettor. Sterile distilled water (1.0 ml) was then added and teliospores vortexed to rinse. The teliospore suspension was again centrifuged for 10 seconds at 1000 X g and supernatant removed with a pipettor. Two additional rinses with sterile distilled water were performed in the same manner.
Following surface sterilization, the teliospores were incubated in 1.0 ml sterile distilled water for 24 hours at 24 C to stimulate production of basidiospores from teliospore surfaces. After 24 hours, basidiospores were dislodged from teliospores by vortexing the 1.5 ml tube for approximately one minute. The basidiospore/teliospore suspension was then left to settle for 5 minutes at 24 C, with heavier teliospores separating from the lighter basidiospores by gravity. After 5 minutes, a clearer upper layer and brown lower layer in the suspension became visible within the suspension, with greater numbers of basidiospores populating the clearer upper layer and teliospores populating the lower layer. 100 ml aliquots of the upper suspension layer were transferred and spread onto 100 mm diameter water agar (1% w/v) plates. The plates were immediately inspected under a stereomicroscope, and ungerminated single basidiospores identified and transferred with a sterile, cool scalpel to Malt Extract Peptone Agar (MPA) medium [1] plates amended with 20 ml/ml ciprofloxacin and 10 ml/ml gentamicin [2].
Mycelium from a single basidiospore (isolate Tm001-NY21) was grown in Malt Extract Peptone Broth (MPB) medium for 14 days at 24°C and shaking at 100 rpm [1]. DNA was extracted with a Macherey-Nagel Nucleobond HMW DNA kit, with the manufacturer’s protocol, with the following modifications: the volumes of lysis buffer H1 and Proteinase K added to the disrupted mycelium in step 2 were increased from 5 ml and 200 ml to 10 ml and 400 ml respectively, and the volume of binding buffer H2 added to the sample in step 6 of the protocol was also doubled from 10 ml to 20 ml. The remainder of the protocol was performed as directed by the manufacturer. Final DNA quality was assessed with a Thermo Scientific Nanodrop and Agilent Femto Pulse System fragment analyzer. A sequence check of the internal transcribed spacer (ITS) region of the genomic DNA was performed using universal fungal ITS PCR primers ITS1 and ITS4 [3] followed by Sanger sequencing and NCBI BLAST analysis (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
Fungal isolation and RNA extraction
For RNA isolation from fungal tissue, mycelium ofTilletia maclaganii Tm001-NY21 was grown in 100 ml MPB medium for 14 days with shaking (50 rpm) at 24°C. Three hours prior to harvest, the fungus was challenged with pentachloronitrobenzene (PCNB) fungicide at a final concentration of 1 ppm. After incubation with PCNB, the mycelium was harvested and filtered, flash-frozen in liquid nitrogen, and disrupted with mortar and pestle. Total RNA was then extracted using a New England Biolabs Monarch Total RNA Miniprep Kit (New England Biolabs, MA, USA) according to the manufacturer's instructions.
Total RNA extraction from switchgrass infected with Tilletia maclaganii was performed by first harvesting infected plant tissue (stems and panicles), flash freezing in liquid nitrogen, then disrupting the plant tissue with mortar and pestle. Approximately 100 mg of disrupted tissue was then processed for total RNA extraction in a manner identical to that described for T. maclaganii Tm001 NY21 mycelium above.
Final RNAs quality was assessed with a Thermo Scientific Nanodrop (Thermo Scientific, MA, USA) and Agilent 5200 Fragment Analyzer (Agilent Technologies, Santa Clara, CA, USA).
DNA sequencing
DNA was sequenced using the PacBio Sequel IIe sequencing platform (Pacific Biosciences, CA, USA). An input of 1.5 µg of genomic DNA was sheared around 10 kb using the Megaruptor 3 (Diagenode, Seraing, Belgium). The sheared DNA underwent exonuclease treatment to remove single-stranded ends. Subsequently, it was subjected to a DNA damage repair enzyme mix and an end-repair/A-tailing mix. The DNA was then ligated with the SMRTbell Barcoded Adapter Plate 3.0 (Pacific Biosciences, CA, USA), using barcoded overhang adapters from the SMRTbell Express Template Prep Kit 2.0 (Pacific Biosciences, CA, USA). Lastly, the DNA was purified using AMPure PB Beads (Pacific Biosciences, CA, USA). Libraries were pooled based on the required amount of reads per sample, ensuring each sample received adequate coverage for analysis. Pooled libraries are then size-selected using the 0.75% agarose gel cassettes with Marker S1 and High Pass protocol on the BluePippin (Sage Science, MA, USA). PacBio Sequencing primer was then annealed to the SMRTbell template library and sequencing polymerase was bound to them using Sequel II Binding kit 2.0.
The prepared SMRTbell template libraries were then sequenced on a PacBio Sequel IIe sequencer using SMRT Link 10.2, 8M v1 SMRT cells, and Version 2.0 sequencing chemistry with 1x1800 movie run times.
RNA sequencing
RNA was sequenced using the Illumina NovaSeq 6000 sequencing platform. Plate-based RNA sample preparation was performed with the PerkinElmer Sciclone NGS robotic liquid handling system, utilizing Illumina's TruSeq Stranded mRNA HT sample prep kit. Poly-A selection of mRNA was carried out following the protocol outlined by Illumina in their user guide (https://support.illumina.com/sequencing/sequencing_kits/truseq-stranded-mrna.html). The PCR conditions were as follows: 1 µg of total RNA starting material per sample and 8 cycles of PCR were used for library amplification. The prepared library was then quantified using KAPA Biosystems next-generation sequencing library qPCR kit (KAPA Biosystem, MA) and run on a Roche LightCycler 480 real-time PCR instrument (Roche, MA, USA).
Once quantified, the library was multiplexed with other libraries, and the pooled libraries sequenced on an Illumina NovaSeq 6000 sequencing platform using NovaSeq XP v1.5 reagent kits, S4 flow cell, following a 2x150 indexed run recipe.
Bioinformatic analysis
Circular consensus sequencing (CCS) reads were initially filtered using the BBMap suite (http://sourceforge.net/projects/bbmap/) to remove low-quality reads, sequencing errors, and other artifacts. The mitochondrial genome was assembled separately from the nuclear genome using CCS reads. Organelle-derived reads were isolated based on coverage and GC content, excluding nuclear sequences using a maximum coverage threshold of 1.5 times the k-mer coverage peak and a GC content cutoff of 0.40 using BBTools version 38.79 [kmercountexact.sh default; bbnorm.sh pigz passes=1 bits=16 target=9999999 min=174; bbduk.sh maxgc=0.4]. An initial mitochondrial assembly was generated with Flye [4] version 2.9-b1768 [-g 100k --asm-coverage 100 --pacbio-hifi], followed by gene prediction using Prodigal [5] version 2.6.3 [-p meta]. Predicted genes were searched against a curated mitochondrial HMM database using HMMER [6] version 3.1b2 [--domtblout]. Contigs with putative mitochondrial genes were masked for ribosomal loci using BBTools [bbduk.sh k=25 mmf=kmask=N] and a curated eukaryotic ribosomal database. These masked contigs were used to recruit additional CCS reads using BBTools [ k=25 mmf= mkf=0.03 ordered ow], and a second Flye assembly was performed with the same parameters. An additional iteration of read recruitment and assembly followed. Contigs shorter than 1 kpb were removed from the assembly and polish was performed with two rounds of Racon [7] version 1.4.13 [racon -u -t 36] to produce the final mitochondrial assembly.
The mitochondrial assembly was used to remove organelle-derived reads from the CCS . The filtered nuclear reads were assembled using Flye version 2.9-b1768 [-t 32 --pacbio-hifi], followed by two additional rounds of Racon [8] polishing.
Genome and mitochondrial assemblies were annotated using the JGI Annotation Pipeline [8-9] with supporting Illumina transcriptome RNA-Seq data.
RNA-Seq reads were filtered using BBduk from the BBmap suite. Raw reads were evaluated for artifact sequences by kmer matching [kmer=25], allowing for 1 mismatch. Detected artifacts were trimmed from the 3' end of the reads. RNA spike-in reads, PhiX reads, and reads containing any N's were removed. Quality trimming was performed using the phred trimming method with a threshold of Q6. After trimming, reads shorter than the length threshold were discarded (minimum length 25 bases or 1/3 of the original read length, whichever was longer). Finally, the trimmed and filtered reads were assembled into consensus sequences using Trinity [10] version 2.11.0.
References
- Eibel P, Wolf GA, Koch E 2005. Detection of Tilletia caries, causal agent of common bunt of wheat, by ELISA and PCR. J. Phytopathology 153:297–306. https://doi.org/10.1111/j.1439-0434.2005.00973.x
- Mondo SJ, Toomer KH, Morton JB, Lekberg Y, Pawlowska TE 2012. Evolutionary stability in a 400-million-year-old heritable facultative mutualism. Evolution 66(8): 2564-2576. https://doi.org/10.1111/j.1558-5646.2012.01611.x
- White TJ, Bruns T, Lee S, Taylor J 1990. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. Pages 315-322 in: PCR Protocols: A Guide to Methods and Applications. M. A. Innis, D. H. Gelfand, J. J. Sninsky, and T. J. White, eds. Academic Press, San Diego, CA.
- Kolmogorov M, Yuan J, Lin Y et al. 2019. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol 37, 540–546 https://doi.org/10.1038/s41587-019-0072-8
- Hyatt D, Chen GL, LoCascio PF et al. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119. https://doi.org/10.1186/1471-2105-11-119
- Mistry J, Finn RD, Eddy SR, Bateman A, Punta M. 2013. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 41(12):e121. https://doi.org/10.1093/nar/gkt263
- Vaser R, Sović I, Nagarajan N, Šikić M. 2017. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27(5):737-746. https://doi.org/10.1101/gr.214270.116
- Kuo A, Bushnell B, Grigoriev IV. 2014. Fungal genomics: sequencing and annotation, p 1–52. In Martin F (ed), Fungi. Advances in botanical research. Elsevier Academic Press, Cambridge, United Kingdom.
- Grigoriev IV, Nikitin R, Haridas S, Kuo A, Ohm R, Otillar R, Riley R, Salamov A, Zhao X, Korzeniewski F, Smirnova T, Nordberg H, Dubchak I, Shabalov I. 2014. MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 42(Database issue):D699-704. https://doi.org/10.1093/nar/gkt1183
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 29(7):644-52. https://doi.org/10.1038/nbt.1883
Protocol references
References
1. Kolmogorov, M., Yuan, J., Lin, Y. et al. 2019. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol 37, 540–546 https://doi.org/10.1038/s41587-019-0072-8
2. Hyatt, D., Chen, G.L., LoCascio, P.F. et al. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119. https://doi.org/10.1186/1471-2105-11-119
3. Mistry J, Finn RD, Eddy SR, Bateman A, Punta M. 2013. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 41(12):e121. https://doi.org/10.1093/nar/gkt263
4. Vaser R, Sović I, Nagarajan N, Šikić M. 2017. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27(5):737-746. https://doi.org/10.1101/gr.214270.116
5. Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 29(7):644-52. https://doi.org/10.1038/nbt.1883
6. Kuo A, Bushnell B, Grigoriev IV. 2014. Fungal genomics: sequencing and annotation, p 1–52. In Martin F (ed), Fungi. Advances in botanical research. Elsevier Academic Press, Cambridge, United Kingdom.
7. Grigoriev IV, Nikitin R, Haridas S, Kuo A, Ohm R, Otillar R, Riley R, Salamov A, Zhao X, Korzeniewski F, Smirnova T, Nordberg H, Dubchak I, Shabalov I. 2014. MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 42(Database issue):D699-704. https://doi.org/10.1093/nar/gkt1183