Mar 18, 2026
CiFi: 3C Library Preparation for PacBio HiFi Sequencing
- Gulhan Kaya1,
- Sean P. McGinty1,
- Mohamed Abuelanin1,
- Megan Y. Dennis1
- 1Genome Center, MIND Institute, and Department of Biochemistry & Molecular Medicine, University of California, Davis, CA 95616, USA
- High molecular weight DNA extraction from all kingdoms
- Dennis Lab_UC Davis Genome Center



Protocol Citation: Gulhan Kaya, Sean P. McGinty, Mohamed Abuelanin, Megan Y. Dennis 2026. CiFi: 3C Library Preparation for PacBio HiFi Sequencing. protocols.io https://dx.doi.org/10.17504/protocols.io.4r3l21zxpg1y/v1
Manuscript citation:
McGinty, S.P., Kaya, G., Sim, S.B. et al. CiFi: accurate long-read chromosome conformation capture with low-input requirements. Nat Commun (2025). https://doi.org/10.1038/s41467-025-66918-y
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: November 05, 2025
Last Modified: March 18, 2026
Protocol Integer ID: 231543
Keywords: 3c library preparation for pacbio hifi, pacbio hifi, preparation of pacbio, pacbio, 3c library preparation, cifi workflow, version of the cifi workflow, cifi, final size selection for hifi, read 3c, hifi
Abstract
This protocol outlines the preparation of PacBio-compatible long-read 3C (CiFi) libraries from crosslinked cells or small tissue samples. The workflow includes low-input handling, restriction digestion, proximity ligation, PCR amplification, SMRTbell library construction, and final size selection for HiFi sequencing.
This protocol represents an amended and updated version of the CiFi workflow compared with the originally published protocol.
Image Attribution
Figures generated by the Dennis Lab (UC Davis Genome Center) for the CiFi method. See associated publication for full experimental context.
Guidelines
- Low-cell inputs: Scale all volumes proportionally (e.g., 50% for 5 M cells; 20% for 1–0.5 M; 10% for 2.5×10⁶–6.25×10⁵).
- Mosquito (An. coluzzii): Minor size-selection adjustments may be required (e.g., double SPRI selection after ligation; 0.45× AMPure PB before SMRTbell; use SMRTbell 3.0 kit).
- Medfly (C. capitata): Fix with 2% formaldehyde; centrifuge at 1500×g for 5 min; reverse crosslinks using Proteinase K/SDS/NaCl mix; perform pre-amplification size selection with 35%-diluted AMPure PB at 3.1× to remove <5 kb fragments; final BluePippin selection >8 kb.
Materials
User Supplied Equipment, Reagents and Consumables:
- 37% Formaldehyde (e.g. Fisher Scientific® Cat # F79-500)
- Glycine (2.5 M stock) for quenching formaldehyde
- 100% Ethanol and freshly prepared 80% and 70% Ethanol
- Phenol:Chloroform:Isoamyl Alcohol (25:24:1 v/v; Thermo Fisher Cat # 15593031)
- 3M sodium acetate pH 5.2 (VWRVE521-100ML)
- Glycogen (optional) (20 µg/µL; Thermo Fisher Cat # 10814010)
- 1X PBS, pH 7.4 (e.g. Fisher Scientific® Cat # 50-842-949)
-Restriction enzyme(s) (e.g., DpnII and/or HindIII)
- T4 DNA Ligase (high concentration)
- Proteinase K
-SDS, Triton X-100, Tween-20, and BSA
- Qubit® Fluorometer, dsDNA HS Assay Kit and consumables (e.g. Thermo Fisher Scientific Cat # 32851, 32856)
- 15mL conical tubes
- 1.7 mL DNA LoBind tubes, PCR tubes, or PCR plates, and a magnetic rack compatible with the selected tubes.
- Centrifuge
- Thermal cycler (if performing Arima-3C Protocol in PCR tubes or PCR plate)
- Thermomixer (if performing Arima-3C Protocol in 1.7mL microcentrifuge tubes)
- Phase Lock Gel tubes
- Wide-bore tips
- Low TE buffer (pH 8.0, Cat# 102-178-400)
Required kits:
- SMRTbell prep kit 3.0, cat# 102-182-700
- Optional: SMRTbell adapter index plate 96A, cat# 102-009-200 (SMRTbell Adapter Index Plate 96A for multiplexing instead of the Overhang Adapter V3)
- SMRTbell cleanup beads, cat# 102-158-300
https://www.pacb.com/wp-content/uploads/Insert-SMRTbell-Cleanup-Beads.pdf
- KOD Xtreme Hot Start DNA Polymerase, Sigma Aldrich cat# 71975-M
- AMPure PB bead size selection kit, cat# PacBio 10-2-182-500
- Twist Universal Adapter System, cat# Twist 101307-101311
Before start
Notes:
- This protocol is an amended and updated version of the CiFi workflow relative to the published protocol.
- See the section “Scaling Down the CiFi Protocol for Lower Cell Inputs” at the end of this protocol.
- Keep samples on ice at all times when not incubating, unless otherwise specified.
- Always use wide-bore tips to minimize DNA shearing.
- The CiFi protocol was derived from and adapted based on previously published methods.
- Chromatin conformation capture (3C) is performed using a Pore-C–based workflow adapted from Deshpande et al. (2022).
- For lymphoblastoid cell lines (LCLs), reagent volumes can be scaled according to input cell number (62,000–10 million cells).
- Modifications have been introduced for non-LCL samples.
- The 3C library is subsequently amplified and converted into SMRTbell libraries using the PacBio ultra-low DNA input protocol, with modifications described by Bein et al. (2024).
- Final size selection is performed using BluePippin (>5 kb) or AMPure PB beads (>3 kb).
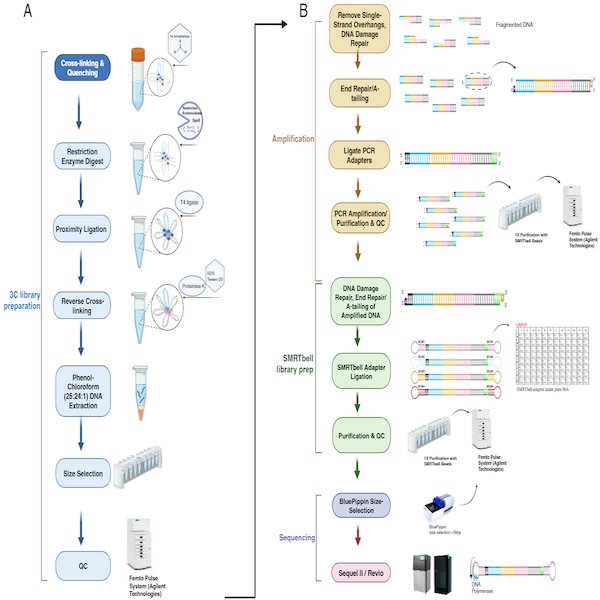
Part 1: 3C Library Preparation
This section describes preparation of the 3C library from crosslinked cells.
- Always use wide-bore tips to minimize DNA shearing
- Ensure reagents are thawed and mixed as directed.
- Avoid overdrying beads during cleanup steps.
- Maintain accurate temperature control during incubations.
- Crosslinked pellets must be free of residual liquid before starting the reaction.
- Use DNA LoBind tubes throughout to minimize sample loss and preserve long DNA fragments.
Step 1 — Crosslink cells
Preparation:
- Wash 5-10 million cells three times in chilled 1X phosphate buffered saline (PBS) in a 50 mL centrifuge tube.
- Centrifuge at 500xg for 5 minutes at 4°C between each wash.
- Resuspend cells in 10 mL room temperature 1X PBS with 1% formaldehyde [EMD Millipore cat no. 818708] by gently pipetting with a wide bore tip.
- Incubate at room temperature for 10 minutes.
Quenching:
- Add 527 µL of 2.5 M glycine (final 125 mM) in 10.5 mL.
- Incubate for 5 minutes at room temperature followed by 10 minutes on ice.
- Pellet the cross-linked cells by centrifugation at 500xg for 5 minutes at 4°C.
- Wash the cross-linked cells with 1x PBS, remove the supernatant, and snap-freeze the pellet in liquid nitrogen. Store at -80°C until you start the protocol.
Step 2 — Restriction Digest
Cell Lysis:
- Resuspend the cell pellet in a mixture of 50 μL of protease inhibitor cocktail [Sigma Aldrich cat no. P8340] in 500 μL of cold permeabilization buffer (10 mM Tris-HCl pH 8.0, 10 mM NaCl, 0.2% IGEPAL CA-630).
- Place on ice for 15 minutes.
- Centrifuge at 500xg for 10 minutes at 4°C.
- Aspirate the supernatant and replace with 200 μL of chilled 1.5X digestion reaction buffer [NEB] compatible with the restriction enzyme used.
- Centrifuge again at 500xg for 10 minutes at 4°C, then aspirate and resuspend in 300 μL of chilled 1.5X digestion reaction buffer.
Chromatin Denaturation:
- Add 33.5 μL of 1% w/v SDS [Thermo Fisher Scientific cat no. 15553027] to each cell suspension.
- Incubate for exactly 10 minutes at 65°C with gentle agitation.
- Place on ice immediately afterwards.
- Quench the SDS by adding 37.5 μL of 10% v/v Triton X-100 [Sigma Aldrich cat no. 93443] for a final concentration of 1%.
- Incubate for 10 minutes on ice.
Digestion:
- Permeabilized cells are then digested with a final concentration of 1 U/μL of DpnII [NEB], brought to volume with nuclease-free water to achieve a final 1X digestion reaction buffer in 450 μL.
- Mix by gentle inversion and incubate in a thermomixer at 37°C for 18 hours with periodic < 1000 rpm rotation (< 30 seconds every 15 minutes) to prevent condensation inside the lid
Step 3 — Proximity Ligation and Reverse Cross-linking
Inactivation:
- DpnII restriction digests are heat-inactivated at 65°C for 20 minutes with 300 rpm rotation. Place on ice immediately afterwards.
Ligation:
- Set up proximity ligation at room temperature with the following reagents:
- 100 μL of 10X T4 DNA ligase buffer [NEB]
- 10 μL of 10 mg/mL BSA
- 50 μL of T4 Ligase [NEB M0202L]
- Total volume of 1000 μL with nuclease-free water.
- Cool to 16°C and incubate for 6 hours with gentle rotation.
Step 4 — Protein Degradation and DNA Purification
Reverse Cross-linking:
- Treat samples with:
- 100 μL 20 mg/mL Proteinase K [Thermo Fisher Scientific cat no. 25530049]
- 100 μL 10% SDS [Thermo Fisher Scientific cat no. 15553027]
- 500 μL 20% v/v Tween-20 [Sigma Aldrich cat no. P9416]
- Total volume of 2000 μL with nuclease-free water.
- Incubate in a thermomixer at 56°C for 18 hours with < 1000 rpm rotation (< 30 seconds every 15 minutes) to prevent condensation inside the lid.
Purification:
Purify DNA using standard phenol-chloroform extraction and ethanol precipitation method.
Phenol–Chloroform Extraction
- Cool the sample on ice after completing the reverse cross-linking incubation (56 °C, ~18 hr).
- Transfer the entire sample (~2.0–2.2 mL) to a 5 mL DNA LoBind centrifuge tube or a 15 mL centrifuge tube.
- Rinse the original tube with 200 µL nuclease-free water and add this to the same 5 mL tube (final volume ~2.2 mL).
- Add an equal volume of chilled phenol:chloroform:isoamyl alcohol (25:24:1)
- Mix thoroughly on a vertical rotator at ~20 rpm for 10 minutes (or invert 20–30 times until fully emulsified).
- Transfer the mixture to 2 mL or 15 mL Phase Lock Gel tubes (recommended for clean phase separation).
- Centrifuge at 16,000 × g, 15 °C for 15 minutes.
- If not using phase-lock tubes:
- Incubate the tube on ice for 2 minutes until the organic layer becomes cloudy to stabilize the interphase.
8. Carefully transfer the aqueous (upper) phase to a new 5 mL DNA LoBind tube or a 15 mL centrifuge tube.
- Avoid disturbing the interphase.
- Expected recovered volume: ~2.0 mL.
Ethanol Precipitation
9. Add 0.1 volumes of 3 M sodium acetate pH 5.2
10. Mix by gentle inversion. The solution may briefly turn cloudy.
11. Add ~3 volumes of 100% ethanol
12. Mix gently by inversion 10–15 times.
13. Precipitate DNA at –80 °C for at least 1 hour or –20 °C overnight (recommended for best recovery).
14. Pre-cool centrifuge to 4 °C.
15. Centrifuge samples at 16,000 × g, 4 °C for 30 minutes.
16. Carefully discard supernatant without disturbing pellet.
17. Wash pellet with 4 mL of 80% ethanol (or 2 mL if using 2 mL tubes).
18. Centrifuge at 16,000 × g, 4 °C for 5 minutes.
19. Discard supernatant, then wash with 2 mL of 70% ethanol.
20. Centrifuge again at 16,000 × g, 4 °C for 5 minutes.
21. Remove supernatant completely. Briefly spin and remove remaining droplets.
22. Air-dry pellet for ~3-5 minutes only.
- Do not overdry; pellet becomes hard to dissolve
23. Resuspend each pellet in 52 µL of TE buffer (pH 8.0).
24. If multiple tubes were used, dissolve the first pellet and transfer the solution onto the next pellet until all are combined.
25. Incubate at room temperature for 5 minutes, gently inverting every few minutes.
26. Briefly spin down.
27. DNA can be pooled into a single 1.5 mL DNA LoBind tube.
28. Store at 4°C short-term or –20°C long-term.
29. Quantify DNA using Qubit DNA HS assay.
30. Proceed to the next CiFi workflow steps (size selection, 3C DNA QC, etc.)
Step 5 —Size Selection
Follow the PacBio protocol for size selection using AMPure PB beads at a 0.45× ratio (to remove fragments smaller than ~3 kb), or alternatively use diluted AMPure PB beads for cleanup and size selection (to remove fragments smaller than ~5 kb)
- For libraries prepared using DpnII, proceed directly to size selection.
- For libraries prepared using HindIII, the longer DNA fragments require an additional shearing step before size selection. Shear the 3C DNA to approximately 10 kb using a g-TUBE (Covaris, 520104), following the manufacturer's protocol.
In summary:
Option#1: Standard PacBio AMPure Beads Cleanup (0.45× Ratio)
1. Add 0.45× volume of AMPure PB beads (Cat# 102-265-900) to your 3C DNA sample. Mix well and incubate for 10 minutes at room temperature.
2. Place the tube on a magnetic rack until the solution clears (~5 minutes). Discard the supernatant carefully.
3. Wash the beads twice with 200 µL of 80% ethanol while keeping the tube on the magnet. Discard ethanol after each wash.
4. Do not overdry. Immediately add 47 µL of Low TE buffer (pH 8.0, Cat# 102-178-400) to elute. Mix well, incubate for 5 minutes, place back on the magnet, and transfer the eluate to a new tube.
Option#2: Diluted AMPure PB bead cleanup and size selection
This step enriches the sample for DNA fragments of the desired size.
Instructions:
Prepare 35% AMPure PB beads by diluting with elution buffer.
- Make a 35% v/v dilution of AMPure PB beads by adding 1.75 mL of resuspended AMPure PB beads to 3.25 mL of elution buffer. The 35% dilution can be stored at 4°C for 30 days
- Add 3.1X volume of diluted beads to the sample.
- Mix thoroughly and spin briefly.
- Let the sample sit for 10 minutes at room temperature to bind DNA.
- Place on a magnetic rack and remove the supernatant.
- Wash beads twice with 200 μL of 80% ethanol.
- Dry beads as before and resuspend in 50 μL of elution buffer.
- Incubate for 5 minutes, then separate beads on the magnet.
- Transfer the supernatant to a new tube for sequencing or storage.
Step 6 — QC
- After size selection, assess library quality by running undigested, digested, and 3C ligation products on a 1% agarose gel. Include a DNA ladder for size reference.
- Successful digestion and ligation typically produce a broad smear corresponding to fragmented and ligated chromatin.
- Optionally, analyze 3C DNA using automated pulsed-field capillary electrophoresis (e.g., Femto Pulse) to confirm the expected fragment size distribution.
- Representative gel images and fragment analyzer profiles are shown below.
Representative agarose gel showing restriction digestion efficiency.
Undigested DNA, enzyme-digested DNA (DpnII or HindIII), and 3C ligation products are shown alongside a 1 kb ladder. Successful digestion produces a smear consistent with fragmented chromatin.
Part 2: Preparing HiFi SMRTbell CiFi Libraries
Notes:
- Skip the gDNA shearing step for DpnII since we already digested it with the restriction enzyme, perform only for HindIII libraries.
- Always use wide-bore tips to prevent damage to the DNA.
- Ensure reagents are thawed and mixed as directed.
I. Amplification
1. Repair and A-Tailing of digested 3C DNA
This step repairs DNA damage and adds A-tails in a single reaction to prepare for adapter ligation.
Repair Master Mix:
- 4 μL Repair Buffer
- 1 μL End Repair Mix
- 0.5 μL DNA Repair Mix
- 24.5 μL Digested 3C DNA Sample
- Total Volume: 30.0 μL
Instructions:
- Mix the Repair Master Mix gently.
- Briefly spin down the mix to collect liquid.
- Add 5.5 µL Repair Master Mix to 24.5 µL DNA (final 30 µL).
- Gently mix the samples and spin briefly to collect contents.
- Set up the thermocycler with the following program:
- 30 minutes at 37°C
- 5 minutes at 65°C
- Hold at 4°C
6. Move to the next step of the protocol.
2. Ligation of Linear Amplification Adapter and Cleanup
This step adds the amplification adapter to DNA fragments. Ensure cleanup beads are at room temperature before starting.
Ligation Master Mix:
- 2 μL Twist universal adapter
- 10 μL Ligation Mix
- 0.5 μL Ligation Enhancer
- 30 μL End-Repaired Sample
- Total Volume: 42.5 μL
Instructions:
- Gently mix the Ligation Master Mix.
- Spin briefly to collect the liquid.
- Add 12.5 µL ligation master mix (Twist universal adapter + ligation reagents) to the 30 µL end-repaired sample (final 42.5 µL).
- Mix samples gently and spin briefly.
5. Use the thermocycler with the following program:
- 30 minutes at 20°C
- Hold at 4°C
Cleanup with 1X SMRTbell Cleanup Beads
- Add 42.5 μL of room-temperature beads to the samples.
- Mix beads with the samples thoroughly.
- Spin briefly to collect liquid.
- Let the samples sit at room temperature for 10 minutes to bind DNA.
- Place samples on a magnetic rack until beads separate.
- Remove the supernatant carefully without disturbing the beads.
- Add 200 μL of 80% ethanol to the beads, wait 30 seconds, and remove ethanol.
- Repeat the ethanol wash step.
- Dry the beads by:
- Removing residual ethanol while on the magnet.
- Briefly spinning and placing back on the magnet.
10. Resuspend the beads in 24 μL of elution buffer.
11. Spin briefly, then incubate for 5 minutes at room temperature.
12. Place samples back on the magnet and transfer the supernatant to a new tube.
3. Amplification and Cleanup
Amplify DNA fragments with adapters on both ends and clean them using 1X SMRTbell Cleanup Beads.
PCR Master Mix:
- 50 μL 2x Xtreme Buffer
- 20 μL dNTPs (2 mM each)
- 4 μL Twist UDI primers (plate)
- 2 μL KOD Xtreme Hot Start DNA Polymerase
- 24 μL Adapter-Ligated Sample
- Total Volume: 100 μL
Instructions:
- Mix the PCR reagents thoroughly.
- Spin briefly to collect the liquid.
- Add 76 μL of PCR mix to each sample, ensuring a final volume of 100 μL.
- Mix samples and spin briefly.
5. Use the thermocycler with this program:
- 94 °C — 2 min (1 cycle)
- 98 °C — 10 sec (10-12 cycles)
- 58.8 °C — 30 sec (10-12 cycles)
- 68 °C — 10 min (10-12 cycles)
- 68 °C — 7 min (1 cycle)
- Hold at 4°C
DNA Input and PCR Cycles:
- 1 ng: 14 cycles
- 5 ng: 12 cycles
- 10 ng: 11 cycles
- 20 ng: 10 cycles
- 50 ng: 8 cycles
Cleanup with 1X SMRTbell Cleanup Beads
- Add 100 μL of beads to the samples.
- Mix thoroughly and spin briefly.
- Let the samples sit for 10 minutes at room temperature to bind DNA.
- Place on a magnetic rack and remove supernatant.
- Wash beads twice with 200 μL of 80% ethanol, waiting 30 seconds each time before removing ethanol.
- Dry beads as before and resuspend in 50 μL of elution buffer.
- Incubate for 5 minutes, then separate beads on the magnet.
- Transfer the supernatant to a new tube for the next step.
II. SMRTbell library prep
4. Repair and A-Tailing of Amplified DNA
This step prepares amplified DNA by repairing damage and adding A-tails for SMRTbell adapter ligation.
- 500-600 ng input or more for Revio or Vega sequencing
- 250 ng input or more for Sequel II/IIe sequencing
Repair Master Mix:
- 8 μL Repair Buffer
- 2 μL End Repair Mix
- 1 μL DNA Repair Mix
- 49 μL Amplified DNA
- Total Volume: 60.0 μL
Instructions:
- Mix the Repair Master Mix gently to combine.
- Spin briefly to ensure all liquid is collected at the bottom.
- Add 11 μL of the mix to each sample for a final volume of 60 μL.
- Mix gently and spin briefly to collect liquid.
- Use the thermocycler with the following program:
- 30 minutes at 37°C
- 5 minutes at 65°C
- Hold at 4°C
6. Proceed to the next step in the workflow.
5. SMRTbell Adapter Ligation and Cleanup
This step ligates the SMRTbell adapter to the ends of the amplified DNA fragments.
Ligation Master Mix:
- 4 μL SMRTbell Adapter*
- 15 μL Ligation Mix
- 1 μL Ligation Enhancer
- 60 μL Repaired Amplified DNA
- Total Volume: 80.0 μL
Instructions:
- Mix the ligation components thoroughly.
- Spin briefly to collect liquid.
- Add the prepared mix to the repaired DNA sample.
- Mix gently and spin briefly.
- Use the thermocycler with this program:
- 30 minutes at 20°C
- Hold at 4°C
Cleanup with 1X SMRTbell Cleanup Beads
- Add 80 μL of cleanup beads to the sample.
- Mix thoroughly and spin briefly.
- Let the sample sit for 10 minutes at room temperature to bind DNA.
- Place on a magnetic rack and remove the supernatant.
- Wash beads twice with 200 μL of 80% ethanol, waiting 30 seconds each time before removing ethanol.
- Dry beads as before and resuspend in 40 μL of elution buffer.
- Incubate for 5 minutes, then separate beads on the magnet.
- Transfer the supernatant to a new tube for the next step.
6. Nuclease Treatment
This step removes un-ligated, semi-ligated, or damaged DNA fragments from the sample.
Nuclease Master Mix:
- 5 μL Nuclease Buffer
- 5 μL Nuclease Mix
- 40 μL SMRTbell Ligated DNA
- Total Volume: 50.0 μL
Instructions:
- Mix the nuclease components thoroughly.
- Spin briefly to collect liquid.
- Add the nuclease mix to the ligated DNA sample.
- Mix gently and spin briefly.
- Use the thermocycler with this program:
- 15 minutes at 37°C
- Hold at 4°C
Cleanup with SMRTbell Cleanup Beads (1X)
- Add 50 μL of cleanup beads to the sample.
- Mix thoroughly and spin briefly.
- Let the sample sit for 10 minutes at room temperature to bind DNA.
- Place on a magnetic rack and remove the supernatant.
- Wash beads twice with 200 μL of 80% ethanol, waiting 30 seconds each time before removing ethanol.
- Dry beads as before and resuspend in 50 μL of elution buffer.
- Incubate for 5 minutes, then separate beads on the magnet.
- Transfer the supernatant to a new tube for the next step.
Size-Selection of SMRTbell Library and Sequencing
Option A: BluePippin high-pass (≥5 kb)
- >5 kb size selection and sequencing on Revio / Sequel II.
or
Option B: 35% diluted AMPure PB method (3.1×, removes <~3–5 kb)
This step enriches the sample for DNA fragments of the desired size.
Instructions:
- Prepare 35% AMPure PB beads by diluting with elution buffer.
- Make a 35% v/v dilution of AMPure PB beads by adding 1.75 mL of resuspended AMPure PB beads to 3.25 mL of elution buffer. The 35% dilution can be stored at 4°C for 30 days
- Add 3.1X volume of diluted beads to the sample.
- Mix thoroughly and spin briefly.
- Let the sample sit for 10 minutes at room temperature to bind DNA.
- Place on a magnetic rack and remove the supernatant.
- Wash beads twice with 200 μL of 80% ethanol.
- Dry beads as before and resuspend in 50 μL of elution buffer.
- Incubate for 5 minutes, then separate beads on the magnet.
- Transfer the supernatant to a new tube for sequencing or storage.
Verification of fragment size during CiFi library preparation. Fragment size distributions were analyzed at key stages of the CiFi library preparation protocol using the Femto Pulse system (Agilent Technologies) for both DpnII (left column) and HindIII (right column) digests. The rows correspond to the following steps in the protocol: (A) 3C DNA QC, (B) After amplification, (C) After SMRTbell library preparation, and (D) After BluePippin size selection.
Reagent Volumes and Procedural Modifications for Low-Input Library Preparation
| A | B | C | D | E | F | |
| Step | 10M cells (original) | 5M cells (50% scale) | 1M-500K cells (20% scale) | 250K-62K cells (10% scale) | C. capitata Mediterranean Fly* | |
| Crosslinking | ||||||
| Wash cells in chilled 1X PBS | 50 mL tube | 50 mL tube | 12 mL tube | 12 mL tube | 1.5 mL tube + 1 mL PBS | |
| Centrifuge at 500xg | 5 min | 5 min | 5 min | 5 min | ||
| Resuspend cells in PBS | 10 mL | 5 mL | 2 mL | 1 mL | ||
| Add 1% formaldehyde | 286 µL | 143 µL | 57.2 µL | 28.6 µL | 142.9 µL of 16% formaldehyde for final concentration of 2% | |
| Incubate at room temp | 10 min | 10 min | 10 min | 10 min | 10 min | |
| Quenching | ||||||
| Add 2.5 M glycine | 527 µL | 263.5 µL | 105.4 µL | 52.7 µL | 130.4 µL | |
| Incubate at room temp | 5 min | 5 min | 5 min | 5 min | 10 min | |
| Incubate on ice | 10 min | 10 min | 10 min | 10 min | ||
| Centrifuge 500xg at 4°C | 5 min | 5 min | 5 min | 5 min | 1500xg for 5 min | |
| Snap-freeze pellet in liquid nitrogen | Store at -80°C | Store at -80°C | Store at -80°C | Store at -80°C | Store at -80°C | |
| Restriction Enzyme Digest | ||||||
| Cell Lysis (Protease inhibitor & buffer) | 550 µL (50 µL + 500 µL) | 275 µL (25 µL + 250 µL) | 110 µL (10 µL + 100 µL) | 55 µL (5 µL + 50 µL) | 550 µL (50 µL + 500 µL) | |
| Incubate on ice | 15 min | 15 min | 15 min | 15 min | 15 min | |
| Centrifuge at 500xg at 4°C | 10 min | 10 min | 10 min | 10 min | 1500xg for 5 min | |
| Add 1.5X digestion buffer | 200 µL | 100 µL | 40 µL | 20 µL | 200 µL | |
| Centrifuge at 500xg at 4°C | 5 min | 5 min | 5 min | 5 min | 1500xg for 5 min | |
| Resuspend in chilled 1.5x digestion buffer | 300 µL | 150 µL | 60 µL | 30 µL | 300 µL | |
| Chromatin Denaturation | ||||||
| Add 1% SDS | 33.5 µL | 16.75 µL | 6.7 µL | 3.3 µL | 33.5 µL | |
| Incubate at 65°C | 10 min | 10 min | 10 min | 5 min | 10 min | |
| Quench with Triton X-100 | 37.5 µL | 18.75 µL | 7.5 µL | 3.75 µL | 37.5 µL | |
| Incubate on ice | 10 min | 10 min | 10 min | 10 min | 10 min | |
| Digestion | ||||||
| Add DpnII & water | 45 µL & 34 µL | 22.5 µL & 17 µL | 9 µL & 6.8 µL | 4.5 µL & 3.4 µL | NlaIII 45 µL & 34 µL; HindIII 22.5 µL & 56.5 µL | |
| Total volume | 450 µL | 225 µL | 90 µL | 45 µL | 450 µL | |
| Incubate at 37°C with rotation | 18 hrs | 18 hrs | 6 hrs | 2 hrs | 16 hrs | |
| Proximity Ligation & Reverse Crosslinking | ||||||
| DpnII inactivation | 65°C, 20 min | 65°C, 20 min | 65°C, 20 min | 65°C, 20 min | 65°C, 20 min | |
| Add ligase buffer, BSA, T4 Ligase, & water | 100 µL, 10 µL, 50 µL, 395 µL | 50 µL, 5 µL, 25 µL, 197.5 µL | 20 µL, 2 µL, 10 µL, 79 µL | 10 µL, 1 µL, 5 µL, 40 µL | 100 µL, 10 µL, 50 µL, 390 µL | |
| Total ligation volume | 1000 µL | 500 µL | 200 µL | 100 µL | 1000 µL | |
| Incubate at 16°C | 6 hrs | 6 hrs | 4 hrs | 4 hrs | 6 hrs | |
| Protein Degradation & DNA Purification | ||||||
| Add Proteinase K, SDS, Tween-20, & water | 100 µL, 100 µL, 500 µL, 300 µL | 50 µL, 50 µL, 250 µL, 150 µL | 20 µL, 20 µL, 100 µL, 60 µL | 10 µL, 10 µL, 50 µL, 30 µL | 100 µL, 100 µL, 0 µL, 728 µL, 72 µL 5M NaCl | |
| Total volume | 2000 µL | 1000 µL | 400 µL | 200 µL | 1000 µL | |
| Incubate at 56°C with rotation | 18 hrs | 18 hrs | 6 hrs | 6 hrs | 65C for 18 hrs at 900 rpm | |
| Purify DNA | Standard phenol-chloroform extraction and ethanol precipitation | Standard procedure | Standard procedure | Standard procedure | 1.8x Ampure bead clean up | |
| Size Selection | ||||||
| Prior to gDNA amplification | Ampure 0.45x | Ampure 0.45x | Ampure 0.45x | Ampure 0.45x | PB Ampure 5 kb cutoff (3.1x, 35% PB Ampure) | |
| After library preparation | Blue pippin 5 kb high pass | Blue pippin 5 kb high pass | Blue pippin 5 kb high pass | Blue pippin 5 kb high pass | Blue pippin 8 kb high pass |