Jul 03, 2026
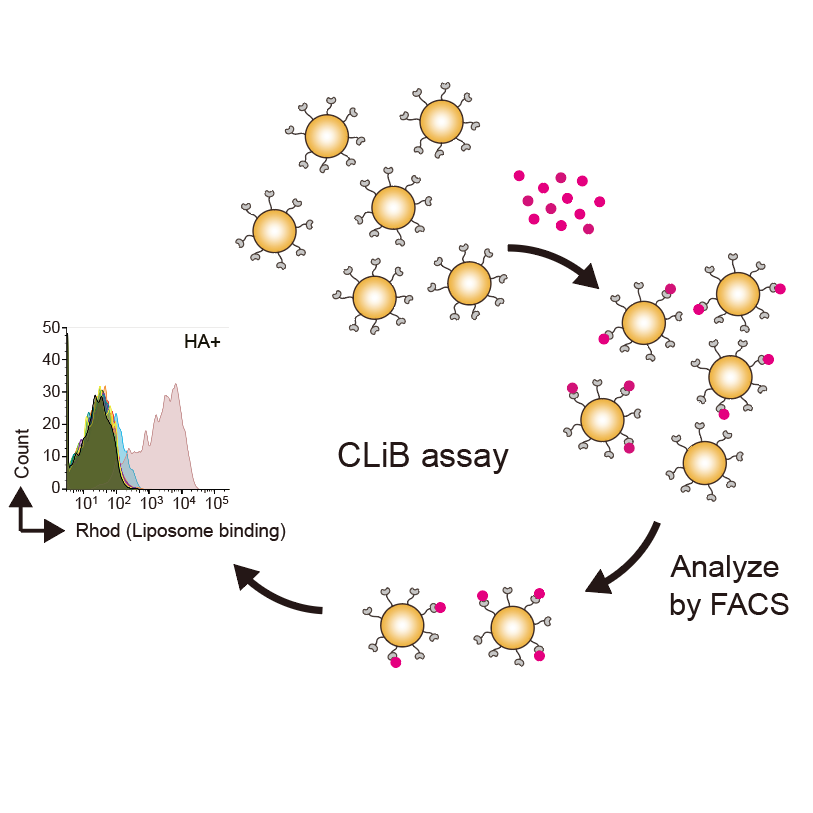
Cell surface Liposome Binding (CLiB) assay
Peer-reviewed method
- Taki Nishimura1,
- Kotaro Tsuboyama2
- 1Institute for Protein Research, The University of Osaka;
- 2Institute of Industrial Science, The University of Tokyo



External link: https://doi.org/10.1038/s41556-026-01996-8
Protocol Citation: Taki Nishimura, Kotaro Tsuboyama 2026. Cell surface Liposome Binding (CLiB) assay. protocols.io https://dx.doi.org/10.17504/protocols.io.14egn5q1qg5d/v1
Manuscript citation:
Nishimura, T., Tsuboyama, K., Nakagaki, Y. et al. Cell surface liposome binding (CLiB) allows lipid-binding probe engineering via high-throughput screening. Nat Cell Biol (2026). https://doi.org/10.1038/s41556-026-01996-8
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: April 22, 2026
Last Modified: July 03, 2026
Protocol Integer ID: 315499
Keywords: CLiB assay, Protein-lipid interaction assay, Lipid-binidng domain
Funders Acknowledgements:
JST PRESTO
Grant ID: JPMJPR20EC
JST PRESTO
Grant ID: JPMJPR21E9
JST FOREST
Grant ID: JPMJFR226A
JST FOREST
Grant ID: JPMJFR230Z
JST CREST
Grant ID: JPMJCR23B6
JST A-STEP
Grant ID: JPMJTR24U7
JST GTeX
Grant ID: JPMJGX23B9
JST GTeX
Grant ID: JPMJGX23B4
JSPS
Grant ID: 25H02268
JSPS
Grant ID: 26H01632
JSPS
Grant ID: 21H05146
JSPS
Grant ID: 24K02019
JSPS
Grant ID: 24H01356
JSPS
Grant ID: 24H01117
The Mishima Kaiun Memorial Foundation
Grant ID: n/a
ONO Medical Research Foundation
Grant ID: n/a
Astellas Foundation for Research on Metabolic Disorders
Grant ID: n/a
The Uehara Memorial Foundation
Grant ID: n/a
The Nakatani Foundation
Grant ID: n/a
Leading Pioneers Science Foundation
Grant ID: n/a
OU Master Plan Implementation Project promoted under Osaka University
Grant ID: n/a
SECOM Science and Technology Foundation
Grant ID: n/a
The Takeda Science Foundation
Grant ID: n/a
The Inoue Foundation for Science
Grant ID: n/a
The Nakajima Foundation
Grant ID: n/a
The Daiichi Sankyo Foundation of Life Science
Grant ID: n/a
The Mitsubishi Foundation
Grant ID: n/a
UTEC-UTokyo FSI Research Grant Program
Grant ID: n/a
The Naito Foundation
Grant ID: n/a
SHIONOGI INFECTIOUS DISEASE RESEARCH PROMOTION FOUNDATION
Grant ID: n/a
Kobayashi Foundation for Cancer Research
Grant ID: n/a
The Asahi Glass Foundation
Grant ID: n/a
AMED-BINDS
Grant ID: JP25ama121016
Disclaimer
This protocol is provided in good faith as a description of the procedures performed by the authors under the conditions described. The authors and their institutions accept no responsibility or liability for any loss, damage, or injury resulting from the use of this protocol. Users are responsible for verifying, optimizing, and applying the protocol at their own discretion and risk. This protocol is intended for research use only and does not constitute medical advice or a guarantee of compliance with local regulations or safety standards.
Abstract
This protocol describes a workflow for the Cell surface Liposome Binding (CLiB) assay to analyze protein–lipid interactions. The procedure covers buffer preparation, construction of yeast display libraries, liposome preparation, flow cytometry–based analysis, and next-generation sequencing (NGS) analysis. Using this workflow, it is possible to rapidly identify and engineer lipid-binding domains with defined specificity and affinity for diverse membrane lipids. The protocol can be applied both to the analysis of lipid-binding domains and to on-demand generation of lipid-binding probes.
Guidelines
This protocol follows standard laboratory safety guidelines for handling biological materials.
Materials
| Reagent | Source | Identifier | |
| DOPE | Avanti | 850725C | |
| DOPC | Avanti | 850375C | |
| Rhodamine-PE (18:1 Liss Rhod PE) | Avanti | 810150C | |
| Anti-HA-tag mAb-Alexa Fluor 647 (TANA2) | MBL | M180-A64 | |
| Yeast Extract | Nacalai | 15838-45 | |
| Bacto peptone | Gibco | 211677 | |
| Yeast nitrogen base without amino acids and ammonium sulfate | BD Difco | 233520 | |
| Ammonium sulfate | Nacalai | 02620-75 | |
| Glucose | Nacalai | 16806-25 | |
| Galactose | Wako | 075-00035 | |
| Drop-out Mix Synthetic, -Trp | USBiological | D9531 | |
| Maltose monohydrate | Nacalai | 21116-05 | |
| BSA | Wako | 015-27053 | |
| Dimethyl Sulfoxide | Nacalai | 08904-14 | |
| Low binding tube | WATSON | PK-15C-500N | |
| JBS Error-prone Kit | Jena Bioscience | PP-102 | |
| Quick taq HS Dye Mix | TOYOBO | DTM-101 | |
| KOD one | TOYOBO | KMM-101 | |
| DpnI | TAKARA | 1235A | |
| NucleoSpin Gel and PCR Clean-up | MACHEREY-NAGEL | 740609.50 | |
| Zymoprep Yeast Plasmid Miniprep I | Zymo Research | D2001 | |
| Zymolase 20T | Nacalai | 07663-91 | |
| Sorbitol | Sigma-Aldrich | 28-4770-5 | |
| Calcium Chloride | Nacalai | 06729-55 | |
| Dithiothreitol | Nacalai | 14112-94 | |
| Lithium Acetate Dihydrate | Wako | 120-01535 | |
| BJ5465 [MATα ura352 trp1 leu2Δ1 his3Δ200 pep4::HIS3 prb1Δ1.6 R can1 GAL] | ATCC | ATCC-208289 | |
| TN1840_pYDS649 plasmid | Addgene | Deposit upon formal acceptance | |
| TN619_fw for vector PCR (GATCCTACCCATACGATGTTCCAGATTA) | Invitrogen | n/a | |
| TN620_rv for vector PCR (TCGACTTCTCTCTTGTCCAATTGAACACCT) | Invitrogen | n/a | |
| TN355_2ndPCR_fw (AACACCACCATCGCTTCTATCGCTGCTAAGGAAGAAGGTGTTCAATTGGACAAGAGAGAAGTCGAC) | Invitrogen | n/a | |
| TN356_2ndPCR_rv (TACTGATGCTTCTGTAGAGGGTGAGGATGTTTGAGCGTAATCTGGAACATCGTATGGGTAGGATCC) | Invitrogen | n/a | |
| KT196_NGS-1stPCR_N4_fw (TCTTTCCCTACACGACGCTCTTCCGATCTNNNNACAAGAGAGAAGTCGACATG) | Invitrogen | n/a | |
| KT200_NGS-1stPCR_N4_rv (GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNTAGGATCCTGATCCACCGCC) | Invitrogen | n/a | |
| P5 (AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCG) | Invitrogen | n/a | |
| P7_8nt_01 (CAAGCAGAAGACGGCATACGAGATgtcggtaaGTGACTGGAGTTCAGACGTGTGCTCTTC) | Invitrogen | n/a | |
| P7_8nt_02 (CAAGCAGAAGACGGCATACGAGATaggtcactGTGACTGGAGTTCAGACGTGTGCTCTTC) | Invitrogen | n/a | |
| P7_8nt_03 (CAAGCAGAAGACGGCATACGAGATgaatccgaGTGACTGGAGTTCAGACGTGTGCTCTTC) | Invitrogen | n/a | |
| P7_8nt_04 (CAAGCAGAAGACGGCATACGAGATgtaccttgGTGACTGGAGTTCAGACGTGTGCTCTTC) | Invitrogen | n/a | |
| P7_8nt_05 (CAAGCAGAAGACGGCATACGAGATcatgaggaGTGACTGGAGTTCAGACGTGTGCTCTTC) | Invitrogen | n/a | |
| P7_8nt_06 (CAAGCAGAAGACGGCATACGAGATtgactgacGTGACTGGAGTTCAGACGTGTGCTCTTC) | Invitrogen | n/a | |
| P7_8nt_07 (CAAGCAGAAGACGGCATACGAGATcgtattcgGTGACTGGAGTTCAGACGTGTGCTCTTC) | Invitrogen | n/a | |
| P7_8nt_08 (CAAGCAGAAGACGGCATACGAGATctcctagaGTGACTGGAGTTCAGACGTGTGCTCTTC) | Invitrogen | n/a | |
| P7_8nt_09 (CAAGCAGAAGACGGCATACGAGATtagttgcgGTGACTGGAGTTCAGACGTGTGCTCTTC) | Invitrogen | n/a | |
| P7_8nt_10 (CAAGCAGAAGACGGCATACGAGATgagatacgGTGACTGGAGTTCAGACGTGTGCTCTTC) | Invitrogen | n/a | |
| P7_8nt_11 (CAAGCAGAAGACGGCATACGAGATaggtgtacGTGACTGGAGTTCAGACGTGTGCTCTTC) | Invitrogen | n/a | |
| P7_8nt_12 (CAAGCAGAAGACGGCATACGAGATtaatgccgGTGACTGGAGTTCAGACGTGTGCTCTTC) | Invitrogen | n/a | |
| P7_8nt_13 (CAAGCAGAAGACGGCATACGAGATtcagacgaGTGACTGGAGTTCAGACGTGTGCTCTTC) | Invitrogen | n/a | |
| P7_8nt_14 (CAAGCAGAAGACGGCATACGAGATgataggctGTGACTGGAGTTCAGACGTGTGCTCTTC) | Invitrogen | n/a | |
| P7_8nt_15 (CAAGCAGAAGACGGCATACGAGATtggtacagGTGACTGGAGTTCAGACGTGTGCTCTTC) | Invitrogen | n/a | |
| P7_8nt_16 (CAAGCAGAAGACGGCATACGAGATcaaggtctGTGACTGGAGTTCAGACGTGTGCTCTTC) | Invitrogen | n/a | |
| P7_8nt_17 (CAAGCAGAAGACGGCATACGAGATgctatcctGTGACTGGAGTTCAGACGTGTGCTCTTC) | Invitrogen | n/a | |
| P7_8nt_18 (CAAGCAGAAGACGGCATACGAGATatggaaggGTGACTGGAGTTCAGACGTGTGCTCTTC) | Invitrogen | n/a | |
| P7_8nt_19 (CAAGCAGAAGACGGCATACGAGATtcaaggacGTGACTGGAGTTCAGACGTGTGCTCTTC) | Invitrogen | n/a | |
| P7_8nt_20 (CAAGCAGAAGACGGCATACGAGATgttacgcaGTGACTGGAGTTCAGACGTGTGCTCTTC) | Invitrogen | n/a | |
| P7_8nt_21 (CAAGCAGAAGACGGCATACGAGATagtctgtgGTGACTGGAGTTCAGACGTGTGCTCTTC) | Invitrogen | n/a | |
| P7_8nt_22 (CAAGCAGAAGACGGCATACGAGATgcacgtaaGTGACTGGAGTTCAGACGTGTGCTCTTC) | Invitrogen | n/a | |
| P7_8nt_23 (CAAGCAGAAGACGGCATACGAGATaaccttggGTGACTGGAGTTCAGACGTGTGCTCTTC) | Invitrogen | n/a | |
| P7_8nt_24 (CAAGCAGAAGACGGCATACGAGATattgcgtgGTGACTGGAGTTCAGACGTGTGCTCTTC) | Invitrogen | n/a |
Safety warnings
Chemical safety
- DMSO: Flammable. Penetrates skin. Wear gloves and dispose as chemical waste.
- LiAc and DTT: Irritant to eyes and skin. Wear gloves and safety glasses.
Equipment safety:
- Always balance centrifuge tubes before use.
GMO safety:
- Handle genetically modified organisms according to institutional biosafety guidelines and dispose of them as biological waste.
Ethics statement
Not applicable.
Before start
- Before starting, verify that all required approvals (biosafety and institutional) have been obtained and that the work will be carried out in an appropriate laboratory environment.
- Prepare all reagents, consumables, and equipment listed in the Materials section.
- Ensure that all stock solutions, culture media, and deionized/distilled water (DDW) are free of contaminants.
- Confirm that the flow cytometer and/or cell sorter is properly calibrated and that appropriate compensation controls and gating strategies have been established.
- Verify that you have access to an Illumina MiSeq or NovaSeq 6000 system for sequencing.
Buffer preparation
17h 35m
YP medium (for 180 mL x 5 bottles)
| Reagent | Amount | |
| Yeast extract (nacalai #15838-45) | 10 g | |
| Bacto peptone (Gibco #211-677) | 20 g | |
| DDW | 900 mL |
- Dissolve all components in DDW with stirring.
- Divide into five 200 mL bottles (180 mL per bottle).
- Autoclave at 121°C for 20 min.
2h 30m
YPD medium (for 200 mL)
| Reagent | Amount | |
| YP medium | 180 mL | |
| 20% glucose | 20 mL |
Mix all components under sterile conditions.
5m
YN medium (for 400 mL x 5 bottles)
| Reagent | Amount | |
| Yeast nitrogen base w/o AA, ammonium sulfate (BD #233520) | 4.25 g | |
| Ammonium sulfate (nacalai, 02620-75) | 12.5 g | |
| 5N NaOH | 1.875 mL | |
| DDW | up to 2000 mL |
- Dissolve all components in DDW with stirring.
- Divide into five 500 mL bottles (400 mL per bottle).
- Autoclave at 121°C for 20 min.
2h 30m
YND-Trp (for 500 mL)
| Reagent | Amount | |
| YN medium | 400 mL | |
| 20% glucose | 50 mL | |
| 10 x AA (-Trp) | 50 mL |
Mix all components under sterile conditions.
5m
YNGal-Trp (for 500 mL)
| Reagent | Amount | |
| YN medium | 400 mL | |
| 20% Galactose | 50 mL | |
| 10x AA (-Trp) | 50 mL |
Mix all components under sterile conditions.
5m
20% glucose
| Reagent | Amount | |
| D-(+)-Glucose (Nacalai, 16806-25) | 100 g | |
| DDW | up to 500 mL |
- Dissolve glucose in DDW with stirring.
- Autoclave at 121°C for 20 min.
2h 30m
20% GAL
| Reagent | Amount | |
| Galactose (Wako, 075-00035) | 100 g | |
| DDW | up to 500 mL |
- Dissolve galacose in DDW with stirring.
- Autoclave at 121°C for 20 min.
2h 30m
10 x AA (-Trp)
| Reagent | Amount | |
| Drop-out Mix Synthetic, -Trp (USBiological, D9531) | 9.9 g | |
| DDW | up to 500 mL |
- Dissolve powder in DDW with stirring and gentle heating.
- Sterilize by filtration through a 0.22 μm filtration.
- Store at 4℃ until use.
1h
Selection B for CLiB assay
| Reagent | Amount | |
| 1M Hepes-NaOH, pH7.5 | 10 mL | |
| 5M NaCl | 15 mL | |
| 50mM Maltose | 50 mL | |
| Albumin, from Bovine Serum [BSA] (Fujifilm, 015-27053) | 0.50 g | |
| DDW | 425 mL |
- Dissolve BSA with stirring.
- Sterilize by filtration through a 0.22 μm filtration.
- Store at 4℃ until use.
30m
50 mM Maltose
| Reagent | Amount | |
| Maltose (nacalai, 21116-05) | 9 g | |
| DDW | up to 500 mL |
- Dissolve maltose in DDW with stirring.
- Sterilize by filtration through a 0.22 μm filtration.
- Store at 4℃ until use.
30m
5N NaOH
| Reagent | Amount | |
| Sodium Hyroxide [nacalai, 31511-05] | 8 g | |
| DDW | up to 40 mL |
- Dissolve sodium hydroxide in DDW with vortexing.
- Cool on ice
- Store at room temperature
5m
1M CaCl2
| Reagent | Amount | |
| Calcium chloride (nacalai 06729-55) | 5.549 g | |
| DDW | up to 50 mL |
- Dissolve calcium chloride in DDW with vortexing.
- Sterilize by filtration through a 0.22 μm filtration.
- Store at room temperature
5m
1M DTT
| Reagent | Amount | |
| Dithiothreitol (nacalai 14112-94) | 2.31 g | |
| DDW | up to 15 mL |
- Dissolve dithiothreitol in DDW with vortexing.
- Sterilize by filtration through a 0.22 μm filtration.
- Aliquot 1 mL into a sterile 1.5 mL tube.
- Store at -30℃ until use.
10m
1M Sorbitol (Glucitol)
| Reagent | Amount | |
| D-Glucitol (nacalai, 06286-55) | 91.085 g | |
| DDW | up to 500 mL |
- Dissolve glucitol in DDW with stirring.
- Autoclave at 121°C for 20 min.
- Store at 4℃ until use.
2h 30m
1M LiAc solution
| Reagent | Amount | |
| Lithium acetate dihydrate (Wako, 120-01535) | 51.01 g | |
| DDW | up to 500 mL |
- Dissolve lithium acetate dihydrate in DDW with stirring.
- Autoclave at 121°C for 20 min.
- Store at room temperature.
2h 30m
Liposome preparation for CLiB assay
1h 10m
Lipids are mixed at the desired molar ratio and the organic solvent is removed under a stream of nitrogen gas, and then further dried using a rotary evaporator (MV-100, TOMY) for 20 min at room temperature.
- Blocking liposome (500 μM): 70 mol% of phosphatidylcholine (PC) and 30 mol% of phosphatidylethanolamine (PE)
- Liposomes containing target lipids (200 μM): 68 mol% of PC, 2 mol% of target lipids, and 29 mol% of PE with 1 mol% of rhodamine-conjugated PE
30m
The lipid film is rehydrated in selection buffer and vortexed for 30 sec. The volume of selection buffer depends on the expreiments; however 600-800 μL is recommended to minimize loss during sonication. Concentrated liposomes can be prepared at this step.
5m
The suspension is sonicated on ice using a tip sonicator (Q500 sonicator) with 10 s ON / 10 s OFF cycles for a total of 10 min at 25% amplitude.
10m
Lipid debris is removed by centrifugation at 177,000 × g for 20 min at 4°C. If concentrated liposomes are prepared, adjust the concentration by adding selection buffer to 500 μM for blocking liposome and 200 μM for target liposomes. The liposomes are stored at 4°C and should be used within a few days.
25m
Cell preparation for CLiB assay
2d
Pre-culture
- Yeast cells are cultured at 30°C for a day in 2 mL of YND-Trp medium in a 15 ml sterilized tube (AS ONE, 34180015D).
1d
GAL Induction
- Add 2 mL of YNGal-Trp medium into a new 15 ml sterilized tube, inoculate with 200 μL of the pre-culture, and incubate at 20°C or 30°C for a 20-28 h.
*Lower temperature (20°C) may improve protein folding for some constructs.
1d
CLiB assay
1h 45m
Cell preparation
- After 20-28 h of culture*, measure the OD600 of cell suspension.
*Incubation longer than 36 h may reduce lipid-binding activity.
- Transfer 300 μL of selection buffer into a low-binding tube (WATSON, PK-15C-500N) and add cells corresponding to 0.5OD (~5×10^6 cells).
- Centrifuged at 3,000 × g for 1 min at 4°C and aspirate the supernatant.
15m
Blocking with unlabeled liposomes
- Add 40 μL of 500 μM unlabeled blocking liposomes (70 mol% of PC and 30 mol% of PE) and incubate for 30 min at 25°C with continuous shaking (800 rpm) using a Thermo BIOSAN TS-100C shaker.
30m
Target lipid binding
- Add 20 μL of 200 μM liposomes containing target lipids and rhodamine-conjugated PE, and incubate for an additional 15 min with shaking (800 rpm) using a Thermo BIOSAN TS-100C shaker.
- Centrifuge at 3,000 × g for 1 min at 4°C, and aspirate the supernatant.
20m
Antibody staining
- Add 10 μL of Alexa 647-conjugated anti-HA antibody (MBL M180-A64, diluted 1:200 in selection buffer) and incubate on ice for 15 min.
- After centrifugation and removal of the supernatant, add 500 μL of the selection buffer and mix with a vortex for 30 sec.
20m
FACS sorting
- Analyze the samples by flow cytometry using a FACSymphonyA1 (BD Biosciences).
- Analyze the data using FlowJo software version 10.10.0.
15m
Error-prone PCR
12h 40m
Error-prone PCR (1st PCR) for insert preparation using JBS Error-prone Kit (Jena Bioscience, PP-102)
| Component | Volume | |
| Appropriate fw primer (10 μM) | 1 μL | |
| Appropriate rv primer (10 μM) | 1 μL | |
| Template plasmid (0.02 μg/μL)* | 1 μL | |
| 10x Reaction B | 1 μL | |
| dNTP Error-prone mix | 0.4 μL | |
| Taq polymerase | 0.2 μL | |
| DDW | 4.4 μL | |
| 10 x Error prone solution | 1 μL |
- Mix all components except 10× Error-prone solution by pipetting.
- Add 1 µL of 10× Error-prone solution.
- Mix gently by pipetting.
PCR using the following cycling program.
| Step | Temperature | Time | Cycles | |
| Denaturation | 94°C | 30 sec | 30* | |
| Annealing | 55°C | 30 sec | 30* | |
| Extension | 72°C | 30 sec | 30* | |
| Final hold | 4°C | Hold | - |
*Optimal concentration and PCR cycle number depends on experimental conditions. Based on the results, the cycle number should be adjusted to achieve the desired mutation rate.
1h
2nd PCR using Quick taq (TOYOBO, DTM-101)
| Component | Volume | |
| fw primer TN355 (10 μM) 5'-AACACCACCATCGCTTCTATCGCTGCTAAGGAAGAAGGTGTTCAATTGGAC AAGAGAGAAGTCGAC-3' | 1 μL | |
| rv primer TN356 (10 μM) 5'-TACTGATGCTTCTGTAGAGGGTGAGGATGTTTGAGCGTAATCTGGAACATC GTATGGGTAGGATCC-3' | 1 μL | |
| 1st PCR product | 4 μL | |
| DDW | 88 μL | |
| 2x Quick taq | 100 μL |
Mix all components by pipetting.
PCR using the following cycling program.
| Step | Temperature | Time | Cycles | |
| Initial denaturation | 94°C | 2 min | 1 | |
| Denaturation | 94°C | 30 sec | 30* | |
| Annealing | 55°C | 30 sec | 30* | |
| Extension | 68°C | 30 sec | 30* | |
| Final hold | 4°C | Hold | - |
- Add 5 μL of DpnI (TAKARA, 1235A) and mix by pipetting.
- Incubate at 37°C for 2 h.
- Incubate at 70°C for 15 min to heat-inactivate DpnI.
*Optimal PCR cycle number depends on experimental conditions.
5h
Vector fragment preparation using KOD-one (TOYOBO, KMM-101).
| Component | Volume | |
| fw primer TN619 (10 μM) 5'-GATCCTACCCATACGATGTTCCAGATTA-3' | 10 μL | |
| rv primer TN620 (10 μM) 5'-TCGACTTCTCTCTTGTCCAATTGAACACCT-3' | 10 μL | |
| Plasmid TN1840 (10 ng/μL)* | 10 μL | |
| DDW | 20 μL | |
| 2x KOD-one enzyme | 50 μL |
Mix all components by pipetting.
PCR using the following cycling program.
| Step | Temperature | Time | Cycles | |
| Initial denaturation | 94°C | 2 min | 1 | |
| Denaturation | 98°C | 10 sec | 30* | |
| Annealing | 55°C | 15 sec | 30* | |
| Extension | 68°C | 240 sec | 30* | |
| Final hold | 4°C | Hold | - |
- Add 5 μL of DpnI (TAKARA, 1235A) and mix by pipetting.
- Incubate at 37°C for 2 h.
- Incubate at 70°C for 15 min to heat-inactivate DpnI.
*Optimal concentration and PCR cycle number depend on experimental conditions.
5h
Clean up PCR products using the NucleoSpin PCR Clean-up Kit (TAKARA, 740609). Load all PCR products for each sample onto a single column and elute with 40 μL of DDW.
40m
Verify band sizes using an MCE-202 MultiNA (Shimadzu) and determine the DNA concentration with NanoDrop Ultra spectrophotometer (Thermo Scientific). At least 8 μg of purfied DNAs for both insert and vector are required for electroporation.
1h
Yeast Transformation by Electroporation
4d 10h 55m
Pre-culture
- Add 5 mL of YPD medium into a sterile 15 mL tube.
- Inoculate with a single colony of BJ5465 cells and incubate at 30°C with shaking overnight.
- Place YND (-Trp) plates at 30°C for 1 day and ensure no visible contamination on plates before use.
1d
Culture expansion
- Measure OD600 of the overnight pre-culture.
- Inoculate 100 mL of YPD medium to an initial OD600 of 0.3 and incubate at 30°C with shaking until OD600 reaches 1.4-1.6 (~5 h).
6h
Wash the cells with DDW twice
- Transfer the culture into two 50 mL tubes (~50 mL each).
- Centrifuge at 2,900 × g for 2 min at r.t.
- Discard the supernatant into a waste flask, add 25 mL of autoclaved DDW to each pellet and resuspend by vortexing.
- Centrifuge at 2,900 × g for 2 min at r.t.
- Discard the supernatant into a waste flask, add 25 mL of autoclaved DDW to each pellet and resuspend by vortexing.
- Centrifuge at 2,900 × g for 2 min at r.t.
- Discard the supernatant into a waste flask.
20m
Wash the cells with Sorbitol/CaCl₂ solution
- Add 25 mL of 1 M sorbitol/1 mM CaCl₂ solution* to each pellet and resuspend by vortexing.
- *Prepare: Mix 25 mL of 1 M sorbitol + 25 µL of 1 M CaCl₂
- Centrifuge at 2,900 × g for 2 min at r.t.
- Discard the supernatant into a waste flask.
10m
LiAc/DTT treatment
- Combine both pellets by adding 10 mL of 0.1 M LiAc/10 mM DTT solution** in a 50 mL tube.
- **Prepare fresh: Mix 9 mL DDW + 1 mL of 1 M LiAc + 100 µL of 1 M DTT.
- Resuspend by vortexing.
- Incubate at 30°C with shaking for 30 min.
- Centrifuge at 2,900 × g for 2 min at r.t.
- Discard the supernatant into a waste flask.
45m
Wash with Sorbitol/CaCl₂ solution
- Add 25 mL of 1 M sorbitol/1 mM CaCl₂ solution and resuspend by vortexing.
- Centrifuge at 2,900 × g for 2 min at r.t.
- Discard the supernatant into a waste flask.
10m
Final wash and resuspension
- Add 25 mL of 1 M sorbitol/1 mM CaCl₂ solution and resuspend by vortexing.
- Centrifuge at 2,900 × g for 2 min at r.t.
- Discard the supernatant.
- Resuspend the pellet in ~500 µL of 1 M sorbitol/1 mM CaCl₂ solution*** to achieve a final volume of ~1 mL.
- ***Prepare: Mix 3 mL of 1 M sorbitol + 3 µL of 1 M CaCl₂, then use ~500 µL.
10m
DNA addition and electroporation
- Transfer 330 µL of cell suspension into a 2 mm electroporation cuvette.
- Add ~8 µg of insert DNA and ~8 µg of vector DNA to the cuvette and mix by pipetting.
- Incubate on ice for 5 min.
- Mix by pipetting, then place the cuvette into the chamber of NEPA Porator (NEPAGENE).
- Electroporation at 2500 V.
10m
Recovery and plating
- Immediately transfer the electroporated cells into a 50 mL tube containing 8 mL of 1:1 mix of 1 M sorbitol:YPD.
- Incubate at 30°C with shaking for 1 h.
- Centrifuge at 2,900 × g for 2 min at r.t.
- Discard the supernatant into a waste flask.
- Resuspend the pellet in 10 mL of YND (-Trp) medium.
- Spread 300 µL of undiluted cell suspension onto ~35 YND (-Trp) plates for library construction.
- Spread 300 µL of each dilution (10-4, 10-5, 10-6, 10-7, 10-8) onto a YND (-Trp) plate.
- Incubate all plates at 30°C for 3 days.
3d
Colony counting and library size estimation
- Count the number of colonies on the serial dilution plates and estimate the total library size based on the dilution plates.
- Example calculation: If 10 colonies appear on the 10-7 dilution plate: 10 × 107 = 1.0 × 108 transformants per plate. Multiply by the total number of library plates to estimate total library size.
10m
Colony collection
- Add 1 mL of sterile DDW onto a library plate and scrape the colonies using a sterile spreader and transfer the suspension into a 50 mL conical tube.
- Add another 1 mL of sterile DDW onto the same plate, scrape again, and pool into the same 50 mL tube.
- Repeat these steps for all library plates (~35 plates).
2h
OD600 measurement and cell concentration adjustment
- Measure OD600 of the pooled cell suspension using a 1:100 dilution in DDW.
- Expected range: OD600 = 80–130 (undiluted)
- Centrifuge the pooled cells at 2,900 × g for 2 min at r.t.
- Discard the supernatant into a waste flask.
- Resuspend the pellet in YND (-Trp) medium to achieve a final concentration of 2.0-8.0 × 109 cells/mL.
30m
Preparation of glycerol stocks
- Aliquot 900 µL of cell suspension into a sterile 1.5 mL tube.
- Add 100 µL of sterile DMSO to each vial and mix by pipetting (final DMSO concentration = 10%). A final concentration of 1.8-7.2 × 109 cells/mL.
- Store at -80°C until use.
30m
[Optional] Generation of yeast display libraries using oligo pools
5h
1st PCR for insert preparation using KOD-one (TOYOBO, KMM-101)
- Solubilize the oligo pools by DDW to make a stock solution (~10 ng/μl).
| Component | Volume | |
| Appropriate fw primer (10 μM) | 1 μL | |
| Appropriate rv primer (10 μM) | 1 μL | |
| 1/10× diluted oligo pools (1 ng/μL) | 2 μL | |
| DDW | 8.5 μL | |
| 2x KOD-one enzyme | 12.5 μL |
Mix all components by pipetting.
Thermal cycling conditions
| Step | Temperature | Time | Cycles | |
| Initial denaturation | 94°C | 2 min | 1 | |
| Denaturation | 98°C | 10 sec | x 16* | |
| Annealing | 55°C | 15 sec | ||
| Extension | 68°C | 30 sec | ||
| Final hold | 4°C | ∞ |
*The number of PCR cycles should be chosen based on a test qPCR run and MultiNA analysis to avoid over-amplification.
1h
Analyze PCR products by agarose gel electrophoresis, then excise the target band and purify it using the NucleoSpin Gel Clean-up Kit (TAKARA, 740609). Elute with 20 μL of DDW.
1h
2nd PCR using KOD-one (TOYOBO, KMM-101)
| Component | Volume | |
| fw primer TN355 (10 μM) 5'-AACACCACCATCGCTTCTATCGCTGCTAAGGAAGAAGGTGTTCAATTGGAC AAGAGAGAAGTCGAC-3' | 8 μL | |
| rv primer TN356 (10 μM) 5'-TACTGATGCTTCTGTAGAGGGTGAGGATGTTTGAGCGTAATCTGGAACATC GTATGGGTAGGATCC-3' | 8 μL | |
| 1/10x diluted 1st PCR product | 8 μL | |
| DDW | 76 μL | |
| 2x KOD-one enzyme | 100 μL |
Mix all components by pipetting.
Thermal cycling conditions
| A | B | C | D | |
| Initial denaturation | 94°C | 2 min | 1 | |
| Denaturation | 98°C | 10 sec | x 9* | |
| Annealing | 55°C | 15 sec | ||
| Extension | 68°C | 30 sec | ||
| Final hold | 4°C | ∞ |
*The number of PCR cycles should be chosen based on a test qPCR run and MultiNA analysis to avoid over-amplification.
1h
Validate the size and single band of PCR product by an MCE-202 MultiNA (Shimadzu). If additional bands and/or smear bands are observed, reduce the PCR cycles.
1h
The PCR products are purified using PCR Clean-up kit (TAKARA, 740609). Load all PCR products into a single column and elute with 40 μL of DDW. Determine the DNA concentration using a NanoDrop Ultra spectrophotometer (Thermo Scientific). At least 8 μg of purfied DNA is required for electroporation.
1h
Cell Sorting for High-Throughput CLiB (HT-CLiB) Assay
4d 9h 50m
Pre-culture of yeast display library
- Thaw a yeast display library stock at room temperature.
- Add *50 µL of tje library stock (0.9-3.6 × 108 cells) to 10 mL of YND-Trp medium in a sterile 50 mL tube.
- *The volume can be adjusted depending on the experimental conditions. It is recommended to use more than a tenfold excess relative to the library size to ensure sufficient coverage of all clones.
- Incubate at 30°C with shaking overnight.
1d
GAL induction
- Add 1 mL of the overnight culture to 10 mL of YNGal-Trp medium in a new sterile 50 mL tube.
- Incubate at *20°C or 30°C with shaking for 20-28 h to induce surface expression.
- *Lower temperature (20°C) may improve protein folding for some constructs.
1d
Cell preparation
- Measure OD600 of the induced culture.
- Transfer cells corresponding to 10 OD600 units (~1 × 108 cells) into a low-binding 1.5 mL tube (WATSON, PK-15C-500N).
- Centrifuge at 3,000 × g for 1 min at 4°C and aspirate the supernatant.
- Wash with 1 mL of selection buffer.
- Centrifuge at 3,000 × g for 1 min at 4°C and aspirate the supernatant.
10m
Blocking with unlabeled liposomes
- Add 400 µL of 500 µM unlabeled blocking liposomes (70 mol% PC, 30 mol% PE) to the cell pellet.
- Resuspend by pipetting and incubate at 25°C for 30 min with continuous shaking at 800 rpm using a Thermo BIOSAN TS-100C shaker.
40m
Target lipid binding
- Add 200 µL of 200 µM liposomes containing target lipids and rhodamine-PE, and mix by pipetting.
- Incubate at 25°C for an additional 15 min with shaking at 800 rpm.
20m
Antibody staining
- Centrifuge at 3,000 × g for 1 min at 4°C and aspirate the supernatant.
- Add 40 µL of Alexa Fluor 647-conjugated anti-HA antibody (MBL M180-A64, diluted 1:200 in selection buffer).
- Resuspend by pipetting and incubate on ice for 15 min.
- Centrifuge at 3,000 × g for 1 min at 4°C and aspirate the supernatant.
- Resuspend the cell pellet in 8 mL of selection buffer.
20m
FACS sorting
- Set up the cell sorter (Sony SH800S) with a 100 µm sorting chip in Ultra Purity mode.
- Sort cells* into four fractions (No, Low, Medium, High) based on rhodamine and Alexa Fluor 647 signal intensities, reflecting lipid-binding activity.
*It is recommended to sort at least 400× the original library diversity to ensure sufficient data for NGS analysis.
8h
Recovery culture
- Centrifuge each sorted fraction at 3,000 × g for 2 min at room temperature.
- Aspirate the supernatant, add 2 mL of YND-Trp medium and mix by vortexing.
- Incubate at 30°C with shaking for 2 days.
2d
Glycerol stock preparation
- Transfer 450 µL of cell suspension from each fraction into a sterile 1.5 mL tube.
- Add 50 µL of sterile DMSO and mix by pipetting (final DMSO concentration = 10%).
- Store at -80°C until use.
10m
Preparation of cell pellet for NGS analysis
- Transfer the remaining cell suspension (~1 mL) from each fraction into a sterile 1.5 mL tube.
- Centrifuge at 3,000 × g for 2 min at room temperature.
- Aspirate the supernatant and store at -30°C until use.
10m
Plasmid DNA extraction from yeast cells using Zymoprep Kit
1h 40m
Preparation of zymolase Solution (5.45 U)
| Reagent | Amount | |
| Zymolase 20T (Nacalai Tesque, 07663-91) | 125 mg | |
| 1.2 M sorbitol | up to 10 mL | |
| 1 M sodium phosphate | 0.1 mL |
Aliquots are stored at -30°C until use.
10m
Zymoprep
- Add 150 µL of Solution 1 (D2004-1-10, Zymo Research) containing 20 µL of zymolase solution (5.45 U) to the yeast pellet.
- Incubate at 37°C for 1 h.
- Add 150 µL of Solution 2 (D2004-2-10, Zymo Research) and mix.
- Add 150 µL of Solution 3 (D2004-3-20, Zymo Research) and mix.
- Centrifuge at 17,700 × g for 2 minutes.
- Transfer the supernatant to a new 1.5 mL tube.
- Add 400 µL of isopropanol and mix thoroughly.
- Centrifuge at 17,700 × g for 8 minutes to pellet DNA.
- Discard the supernatant.
- Centrifuge again at 17,700 × g for 2 minutes and remove any residual supernatant.
- Resuspend the pellet in 35 µL of DDW.
- Stored at -30°C until use.
1h 30m
Next generation sequencing (NGS) analysis
1d 6h 45m
Perform a preliminary qPCR to determine the cycle number for the first PCR.
- Use primers that contain sequences complementary to the common region of the plasmid and partial Illumina P5/P7 adapter sequences (ex) KT196, KT200
- Use appropirate NGS-1st PCR primers that contain sequences complementary to the insert region. In summary, the sequence starting at the N4 region (NNNN) to the 3' end of KT196 and KT200 should be replaced with the appropriate sequences.
- Prepare the following 20 µL PCR reaction for each sample.
| Component | Volume | |
| SsoAdvanced Universal SYBR Green Supermix, 2× (Bio-Rad #172-5271) | 10 µL | |
| NGS-1st PCR primer_fw (50 µM) (ex) KT196 | 0.25 µL | |
| NGS-1st PCR primer_rv (50 µM) (ex) KT200 | 0.25 µL | |
| Diluted yeast prep sample (1/10x, 1/30x or 1/100x) | 2 µL | |
| Molecular-grade water | 8.5 µL |
Run qPCR using the following cycling program.
| Step | Temperature | Time | Cycles | |
| Initial denaturation | 95°C | 30 s | 1 | |
| Denaturation | 95°C | 10 s | 40 | |
| Annealing | 55°C | 30 s | 40 | |
| Extension, with plate read | 72°C | 1 min | 40 |
Determine the cycle number for the first PCR from the qPCR amplification curve.
- Use the cycle number at which the amplification efficiency is approximately 50%, in order to avoid over-amplification.
1h
Perform the first PCR using KOD One PCR Master Mix.
- Use primers that contain sequences complementary to the common region of the plasmid and partial Illumina P5/P7 adapter sequences (ex) KT196, KT200
- Use appropirate NGS-1st PCR primers that contain sequences complementary to the insert region. In summary, the sequence starting at the N4 region (NNNN) to the 3' end of KT196 and KT200 should be replaced with the appropriate sequences.
- Prepare the following 25 µL PCR reaction for each sample.
| Component | Volume | |
| KOD One PCR Master Mix, 2× | 12.5 µL | |
| NGS-1st PCR primer_fw (10 µM) (ex) KT196 | 1 µL | |
| NGS-1st PCR primer_rv (10 µM) (ex) KT200 | 1 µL | |
| Diluted yeast prep sample (As determined by qPCR) | 2 µL | |
| Molecular-grade water | 8.5 µL |
Run PCR using the following cycling program.
| Step | Temperature | Time | Cycles | |
| Initial denaturation | 94°C | 2 min | 1 | |
| Denaturation | 98°C | 10 s | As determined by qPCR | |
| Annealing | 55°C | 15 s | As determined by qPCR | |
| Extension | 68°C | 20 s | As determined by qPCR | |
| Hold | 4°C | Hold | 1 |
Keep the first PCR products on ice until further use.
Validate the size and single band of PCR product by an MCE-202 MultiNA (Shimadzu). If additional bands and/or smear bands are observed, reduce the PCR cycles.
1h
Purify the first PCR products using Sera-Mag magnetic beads (Cytiva #29343052).
- Use a 0.9× bead-to-sample volume ratio. (For a 40 µL PCR reaction, add 36 µL of Sera-Mag bead suspension.)
- Mix thoroughly by pipetting, incubate to allow DNA binding, and place the sample on a magnetic stand until the solution becomes clear.
- Remove the supernatant without disturbing the beads.
- Wash the beads twice with freshly prepared 80% ethanol while the tube remains on the magnetic stand.
- Remove residual ethanol and briefly air-dry the beads (~30s). Do not over-dry the beads.
- Elute DNA in molecular-grade water or elution buffer, then transfer the eluate to a new tube.
30m
Use the purified first PCR product as the template for a second preliminary qPCR.
- Use a 50-fold dilution of each first PCR product.
- Prepare the following 20 µL PCR reaction for each sample.
- The P5 primer is common to all samples, whereas an appropriate P7 primer is selected from the list of P7_8nt for each sample.
- P5 primer: AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCG
- P7 primer example (with 8nt barcode): CAAGCAGAAGACGGCATACGAGATgtcggtaaGTGACTGGAGTTCAGACGTGTGCTCTTC
| Component | Volume | |
| SsoAdvanced Universal SYBR Green Supermix, 2× | 10 µL | |
| P5 primer (50 µM) --- common | 0.25 µL | |
| P7_8nt_primer (50 µM) --- use an appropriate one | 0.25 µL | |
| Diluted, purified first PCR product (1/50x) | 1 µL | |
| Molecular-grade water | 8.5 µL |
Run qPCR using the following cycling program.
| Step | Temperature | Time | Cycles | |
| Initial denaturation | 95°C | 30 s | 1 | |
| Denaturation | 95°C | 10 s | 40 | |
| Annealing | 55°C | 30 s | 40 | |
| Extension, with plate read | 72°C | 1 min | 40 |
Determine the cycle number for the second PCR from the qPCR amplification curve.
- Use the cycle number at which the amplification efficiency is approximately 50%, in order to avoid over-amplification.
1h
Perform the second PCR using the purified first PCR product as the template.
- This PCR adds complete Illumina P5 and P7 adapter sequences, including sequencing primer binding regions and sample index sequences.
- The P5 primer is common to all samples, whereas an appropriate P7 primer is selected from the list of P7_8nt for each sample.
- Prepare the following 40 µL PCR reaction for each sample.
| Component | Volume | |
| KOD One PCR Master Mix, 2× | 20 µL | |
| P5 primer (50 µM) ---- common | 0.5 µL | |
| P7_primer (5 µM) --- use an appropriate one | 5 µL | |
| Diluted, purified first PCR product (1/50x) | 2 µL | |
| Molecular-grade water | 12.5 µL |
Run PCR using the following cycling program.
| Step | Temperature | Time | Cycles | |
| Denaturation | 98°C | 10 s | As determined by qPCR | |
| Annealing | 55°C | 5 s | As determined by qPCR | |
| Extension | 68°C | 5 s | As determined by qPCR | |
| Final extension | 68°C | 60 s | 1 | |
| Hold | 4°C | Hold | 1 |
Keep the amplified libraries on ice until purification.
Validate the size and single band of PCR product by an MCE-202 MultiNA (Shimadzu). If additional bands and/or smear bands are observed, reduce the PCR cycles.
1h
Purify the second PCR products using Sera-Mag magnetic beads.
- Use a 0.9× bead-to-sample volume ratio. For a 40 µL PCR reaction, add 36 µL of Sera-Mag bead suspension.
- Mix thoroughly by pipetting, incubate to allow DNA binding, and place the sample on a magnetic stand until the solution becomes clear.
- Remove the supernatant without disturbing the beads.
- Wash the beads twice with freshly prepared 80% ethanol while the tube or plate remains on the magnetic stand.
- Remove residual ethanol and briefly air-dry the beads. Do not over-dry the beads.
- Elute DNA in molecular-grade water or elution buffer, then transfer the eluate to a new tube or plate.
30m
Measure the DNA concentration of each purified library using a Qubit fluorometer (Thermo Fisher Scientific).
- Record the concentration of each sample.
- Use the measured concentrations to calculate the amount of each library required for pooling.
45m
Sequence the pooled library using an Illumina MiSeq or NovaSeq 6000 system (Illumina).
1d
Calculating binding scores from next-generation sequencing data
- Obtain raw NGS read counts for each unique sequence in each sample.
- Using these values, calculate the binding score of each clone from the NGS read counts according to formula X:
1h
Acknowledgements
This study was supported by PRESTO (JPMJPR20EC to T.N. and JPMJPR21E9 to K.T.), FOREST (JPMJFR226A to T.N. and JPMJFR230Z to K.T.), CREST (JPMJCR23B6 to K.T.), A-STEP (JPMJTR24U7 to T.N. and K.T.), and GteX (JPMJGX23B9 to K.T. and JPMJGX23B4 to K.T.) from Japan Science and Technology (JST), Grant-in-Aid for Transformative Research Areas (A) (25H02268, 26H01632 to T.N.), a grant-in-aid for Transformative Research Areas (B) (21H05146 to T.N.), KAKENHI (24K02019 to T.N., 24H01356 to K.T., and 24H01117 to K.T.) from the Japan Society for the Promotion of Science (JSPS), grants from the Mishima Kaiun Memorial Foundation (to T.N.), ONO Medical Research Foundation (to T.N.), Astellas Foundation for Research on Metabolic Disorders (to T.N.), the Uehara Memorial Foundation (to T.N. and to K.T.), the Nakatani Foundation (to T.N. and to K.T.), Leading Pioneers Science Foundation (to T.N. and to K.T.), OU Master Plan Implementation Project promoted under Osaka University (to T.N.), SECOM Science and Technology Foundation (to K.T.), the Takeda Science Foundation (to K.T.), the Inoue Foundation for Science (to K.T.), the Nakajima Foundation (to K.T.), the Daiichi Sankyo Foundation of Life Science (to K.T.), the Mitsubishi Foundation (to K.T.), UTEC-UTokyo FSI Research Grant Program (to K.T.), the Naito Foundation (to K.T.), SHIONOGI INFECTIOUS DISEASE RESEARCH PROMOTION FOUNDATION (to K.T.), Kobayashi Foundation for Cancer Research (to K.T.), the Asahi Glass Foundation (to K.T.), and Research Support Project for Life Science and Drug Discovery (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Number (JP25ama121016).