Jan 16, 2026
Catalyzed reporter deposition fluorescence in situ hybridization (CARD-FISH)
- 1Florida Atlantic University, Marine Biological Laboratory;
- 2Marine Biological Laboratory



Protocol Citation: Molly A Moynihan, Eleanor Greene 2026. Catalyzed reporter deposition fluorescence in situ hybridization (CARD-FISH). protocols.io https://dx.doi.org/10.17504/protocols.io.rm7vzjebrlx1/v1
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
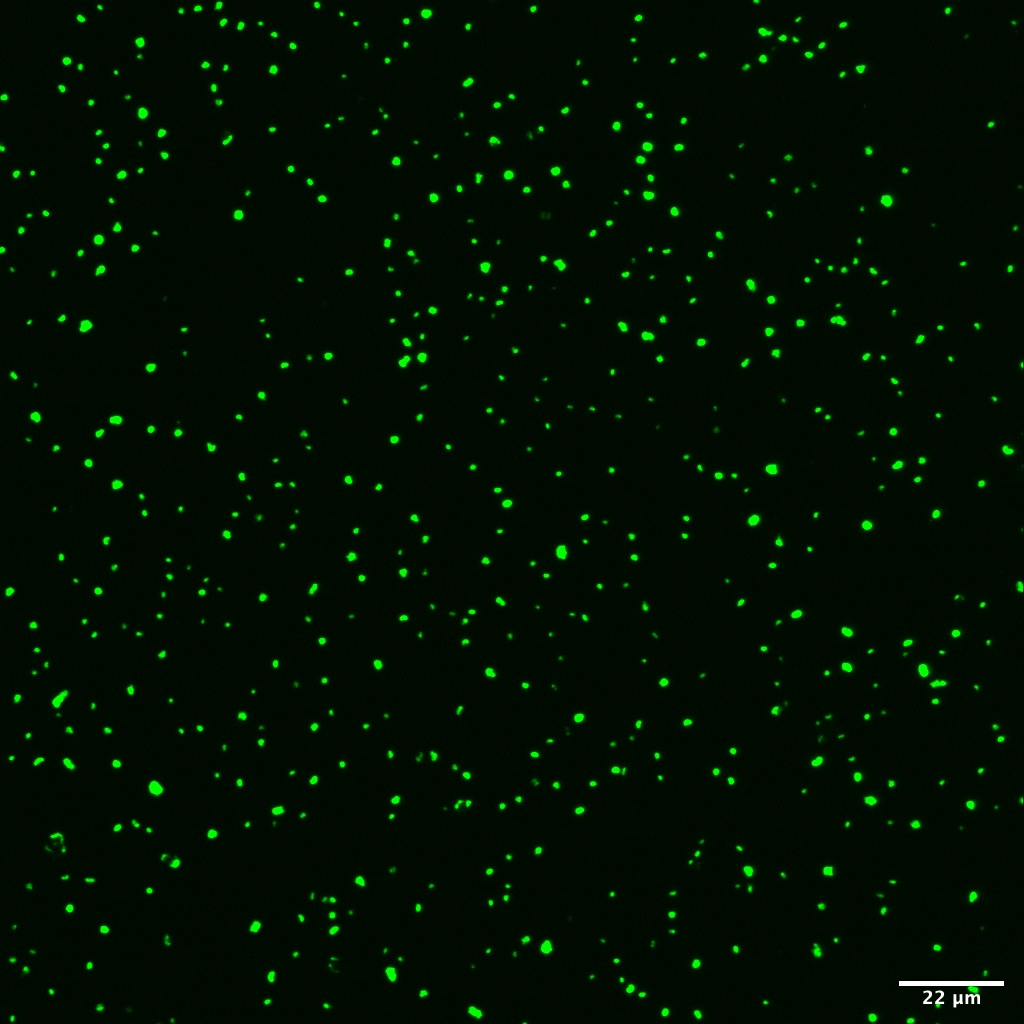
We use this protocol for samples on filters and it's working
Created: August 30, 2024
Last Modified: January 16, 2026
Protocol Integer ID: 106698
Keywords: CARD-FISH, fluorescence in situ hybridization, cell enumeration, DAPI, bacterial cell visualization, catalyzed reporter deposition fluorescence in situ hybridization, catalyzed reporter deposition fluorescence, protocol for catalyzed reporter deposition fluorescence, gammaproteobacteria, fish probe, eubacteria, cells on filter, total cell count, nucleic acid, cell, situ hybridization, methods for dapi
Funders Acknowledgements:
Simons Foundation
Grant ID: 824763
National Science Foundation
Grant ID: 2205993
Abstract
This is a protocol for CAtalyzed Reporter Deposition Fluorescence In Situ Hybridization (CARD-FISH) based on Pernthaler et al. 2002, with modifications from Ishii et al. 2004, Teira et al. 2004, Herndl Lab's protocol, Ruff et al. 2013... see reference list for more details. This protocol has been used successfully for bacterial cell visualization and enumeration of fixed cells on filters with the following CARD-FISH probes: EUB338 I-III (Eubacteria), GSB532 (Chlorobi), and GAM42a (Gammaproteobacteria). The protocol also includes methods for DAPI nucleic acid staining for total cell counts. A detailed list of all materials and catalog numbers is provided in the Materials section.
Materials
Reagents:
- 70% Ethanol
- 80% Ethanol
- 96% Ethanol
- Sterile 1X PBS
- Sterile 10X PBS
- Sterile MilliQ Water
- Low-melting Agarose powder (Fisher Scientific, Cat. No. PR-V2111)
- 37% Hydrochloric Acid (Fisher Scientific, Cat. No. A481-212)
- 20 kU Lysozyme Powder (Fisher Scientific, Cat. No. PI89833) (or 100kU Lysozyme Powder, if available)
- 0.5 M Ethylenediaminetetraacetic Acid (EDTA), pH 8.0 (Fisher Scientific, Cat. No. O2793-500)
- 1 M Tris-HCl, pH 7.5 (Fisher Scientific, Cat. No. PR-H5121)
- 5 M Sodium Chloride (NaCl) (Fisher Scientific, Cat. No. S271-500)
- 20% Sodium Dodecyl Sulfate (SDS) (Fisher Scientific, Cat. No. S529-500)
- 3% or 30% Hydrogen Peroxide (Fisher Scientific, Cat. No. BP2633500)
- Formamide (Fisher Scientific, Cat. no. AC181090010)
- Dextran Sulfate (Fisher Scientific, Cat. No. BP1585-100)
- 5 M Calcium Chloride (Fisher Scientific, Cat. No. AC206795000)
- 1 M Sodium Bicarbonate (Fisher Scientific, Cat. No. BP328-500)
- Blocking Reagent (Roche, Mannheim, Germany, Cat. No. 11 096 176 001)
- Maleic Acid (Fisher Scientific, Cat. No. AC125230010)
- HRP-labeled oligonucleotide probes (50 ng/uL) (Biomers.net)
- Note: Probe aliquots of 4 uL should be made and stored at -20 C. Probes should not be frozen and thawed multiple times. Once the probe has been taken out of the freezer, store at 4 C. However, it's really best practice to just use the probes once after being defrosted.
- Tyramide Working Solution (150 uL DMSO added to stock solution to make 100X working solution)
- Alexa Fluor 594 (Fisher Scientific, Cat. No. B40957)
- Alexa Fluor 488 (Fisher Scientific, Cat. No. B40953)
- Alexa Fluor 647 (Fisher Scientific, Cat. No. B40958)
- Citifluor:Vectashield mixture in a 4:1 ratio (Fisher Scientific, Cat. No. 50-302-34; Fisher Scientific, Cat. No. NC9265087)
- 1X 4',6-diamidino-2-phenylindole (DAPI) (Fisher Scientific, Cat. No. EN62248)
- 2-4% Paraformaldehyde (PFA) for fixation (made from 37% stock of Formaldehyde in 1X PBS, Fisher AC410731000)
Supplies:
- Pencil
- Petri dishes
- Small Spray bottle
- Tweezers
- Whatman paper
- 2 mL microcentrifuge tubes
- 50 mL Falcon tubes
- 15 mL Falcon tubes
- scalpel
- Al foil
- Kim wipes
Stock Solution Preparation (Bulk, prepared ahead of experiment)
Maleic Acid Buffer:
100mM maleic acid, 150 mM NaCl (pH 7.5)
For 500mL, combine:
- 5.805 g Maleic Acid
- 15 mL 5 M NaCl
- DI water to a final volume of 500mL
Adjust to pH 7.5, and then autoclave. Maleic acid buffer can be made ahead of time and kept at room temperature.
Blocking Solution:
10% w/v blocking reagent in maleic acid buffer
For 10mL of blocking solution, combine:
- 1g Blocking Reagent (Roche, Mannheim, Germany, Cat. No. 11 096 176 001)
- 10mL Maleic Acid buffer
Add blocking reagent to maleic acid buffer to faciliate dissolution. Can be made ahead of time and stored at -20°C.
Hybridization Buffer:
0.9M NaCl, 20mM Tris-HCl [pH 7.5], 10% w/v dextran sulfate, 0.02% w/v SDS, 1% w/v blocking reagent, 0-50% formamide, (0.16 ng/ul probe, added on day of experiment, not in stock)
Hybridization buffer can be made in bulk ahead of time at (a) 50% and (b) 0% formamide for storage and then specific formamide concentrations ranging from 0-50% can be made by combining these two in various proportions. Probes are added on the day of an experiment, not in this buffer stock prep.
To make 40mL of (a) 50% and (b) 0% formamide stock hybridization buffers, combine:
- 7.2 mL 5 M NaCl
- 0.8 mL 1 M Tris-HCl, pH 7.5
- 4 mL blocking solution (see above)
- (a) 20 mL formamide for 50% hybridization buffer or
(b) 20 mL of sterile milliQ H2O for 0% hybridization buffer
- 8 mL sterile milliQ
- 4g dextran sulfate
- **40µl 20% w/v sodium dodecyl sulfate (SDS) ** add last to prevent precipitation
Combine the following and then heat at 40-60°C until dextran sulfate has dissolved. This buffer can be pre-made, aliquoted into sterile 2 mL tubes, and stored at -20 C until use.
Washing Buffer:
5mM EDTA [pH 8.0], 20mM Tris-HCl [pH 7.5], 0.01% w/v SDS, 0.9-0.014M NaCl (dependent on formamide concentration)
The NaCl concentration needed in the washing buffer corresponds with formamide concentration in hybridization buffer following the table below. Formamide concentrations are dependent on the probe being used. Washing buffer can be made in bulk at various NaCl concentrations and stored at room temperature.
For 50mL final volume of wash buffer, combine:
- 0.5 mL 0.5 M EDTA, pH 8.0
- 1 mL 1 M Tris-HCl, pH 7.5
- x µL 5 M NaCl (e.g. formamide 10% = 4500 µl; formamide 20% = 2150 µl; formamide 35% =700 µl)
- sterile milliQ H2O to a final volume of 50 mL
- **25 µl 20% w/v sodium dodecyl sulfate (SDS)**add last to prevent precipitation
Amplification Buffer:
2M NaCl, 0.1% w/v blocking reagent, 10% w/v dextran sulfate, 1xPBS
(0.0015% H2O2 and 1:500 parts tyramide, added on day of experiment, not in stock)
To make 40 mL stock amplification buffer, combine:
- 4 mL of sterile 10X PBS, pH 7.4
- 0.4 mL blocking solution (see above)
- 16 mL 5M NaCl
- 4 g dextran sulfate
- sterile milliQ H20 to 40 mL
Heat at 40-60°C until the dextran sulfate has dissolved. Aliquot 1 mL into 2 mL sterile microcentrifuge tubes. When aliquoting, make sure the stock is well combined. Vortex the 50 mL tube after every 5-10 aliquots as needed. Store aliquots at -20°C. H2O2 and tyramide are added to the amplification buffer on the day of use.
Tyramide working solution, 100x
Following manufacturer instructions, add 150µl DMSO to stock solution to make 100x working solution. Make aliquots of 10-20µl. Store aliquots at -20°C. (Freeze/thawing of tyramide working solution is okay).
Tyramides should be chosen based on microscope capabilities and sample type (i.e. avoiding wavelengths that would be autofluorescent. We often use Alexa Fluor 488.)
Probe working solutions, 50ng/ul
Dilute to 50ng/µl and aliquot into 10µl aliquots. Avoid freeze/thaw cycles and this can damage horseradish peroxidase (HRP) enzyme. Store aliquots at -20°C, thaw single 10µl aliquots, store at 4°C for 4-6 months and discard if not used within 6 months of thawing. Do no re-freeze once thawed. Note that each probe has an optimal formamide concentration for hybridization that typically found in the literature specific to that probe.
HRP probes for CARD-FISH can be ordered from Biomers.net
Sample Fixation and Preparation (Critical Step!)
Most sample types should be fixed with 2% paraformaldehyde (PFA) for 8h at 4°C or 1h at room temperature. Thicker samples (e.g. microbial mats, animal tissue) may require up to 4% PFA fixation and increased time, followed by dehydration, embedding and sectioning. Note that fixing the cells for an extended time in PFA (i.e. over-fixation) will prevent the HRP-probe from being able to enter cells. Optimization may be required for various sample types.
Note that 2-4% PFA can be made from 37% stock formaldehyde in 1X PBS.
For planktonic samples, fix with 2% PFA for 8h at 4°C or 1h at room temperature. Samples can either be fixed and then (1) directly filtered onto 0.2µm polycarbonate filters using a 0.45µm cellulose nitrate support filter and at a pressure of approximately 200mbar, rinsed with 2x with 1xPBS to remove PFA, air dried and stored at -20°C. Or, fixed and then (2) centrifuged at 16,000xg for 5 minutes, after which the PFA supernatant is discarded and samples are resuspended in 1x PBS and rinsed 2x with 1x PBS by centrifugation (16,000xg for 5 minutes). Samples can then be stored in 1:1 PBS:EtOH at -20°C and filtered at a later time.
For sediment samples, fix sediment samples in 2-4% PFA for 8h at 4°C or 1h at room temperature. Samples should be fixed in a volume of PFA that is ~10x the sediment volume. After fixation, centrifuge sediment samples at 16,000xg for 5 minutes, discard supernatant and resuspend in 1x PBS. Repeat centrifugation and PBS rinse 2x. Store sediment sample in 1:1 PBS:EtOH at -20°C. Before analysis, sediment samples may need sonication with a sonicating probe. A typical protocol would be: sonicate fixed sediment sample on ice for 30sec at 1 pulse/sec with an intensity of 20%, followed by a 30sec break, repeat 5x. After sonication, filter supernatant onto 0.2µm polycarbonate filters, as described above, using a support filter.
Appropriate filtration volumes for both planktonic and sediment samples should be determined such that there is a countable amount of cells on the filter. In practice, this means filtering samples in several different dilutions (e.g. 10x, 100x, 1000x) and then performing DAPI staining (see below) to look at cell density. Keep track of volumes of sample and dilution factors used in all steps. Also keep track of the diameter of the filter tower used during filtration, as this will be necessary to calculate cell densities.
**Note that when centrifugation is involved in sample prep, some cells will be lost. Thus, for accurate cell enumeration, some of the original sample fixed in 2% PFA (non-washed) must be saved.** This sample can be used for DAPI staining and total cell counting, and compared with DAPI total cell counts of the washed sample. These two numbers are then later used as a correction factor for accurate cell enumeration.
Polycarbonate Filters (GTTP02500): https://www.fishersci.com/shop/products/isopore-membrane-filters-0-2-m/GTTP02500
Cellulose Nitrate Support Filters (14-555-400): https://www.fishersci.com/shop/products/sartorius-cn-membrane-filters-36/14555400#sartorius%20cellulose%20nitrate%20filter
Agarose Sample Embedding (for filter samples)
Note: Depending on the experimental design, whole filters can be embedded with agarose and permeabilized, or filters can be sliced into wedges and only desired filter slices treated with agarose and/or permeabilizied.
If slicing filters into different wedges for different combinations of probes/treatments, be sure to number or label each filter slice accordingly using a pencil. Label only the outer edge of the filter that has no cells (i.e. was not in contact with the sample during filtration).
To make filter slices, use a scalpel and tweezers. Slice a wedge of the filter and place onto a petri dish lined with Whatman paper. Repeat as need for desired number of probe/sample combinations. Make sure to only pick up the filter by the outer edge to avoid contacting the area of the filter with cells.
Prepare a 0.1% w/v solution of low-melting agarose and bring to a boil. Let cool to 30-40°C before use (warm to touch). Solution can be prepared ahead of time and reheated prior to use.
Pour agarose into a spray bottle, and spray the agarose gently onto the cell side of the filter slices for a light coating. Let samples air dry on Whatman paper. If embedding whole filters and then cutting into slices, any remaining agarose embedded filter can be dried and stored at -20°C.
Start of CARD-FISH: Permeabilization and Inactivation with lysozyme (Bacteria)
1h 10m
Prepare permeabilization solutions
Depending on the cell type and environment, various permeabilization solutions can be used. For most bacteria, lysozyme permeabilization described below is sufficient. For Archaea, achromopeptidase, proteinase K and/or HCl treatment may be necessary. **Permeabilization is a critical step for the large HRP-probes to be able to enter the cells.** Optimization of this step is sometimes needed for different cell types.
Lysozyme Permeabilization Solution:
1000 kU/ml lysozyme, 0.05M EDTA [pH 8.0], 0.1M Tris-HCl [pH 7.5]
To make 5mL, combine:
- 50 mg of 100 kU (or 250 mg of 20 kU) lysozyme powder (powder should be stored at -20°C)
- 500 µL 0.5 M EDTA, pH 8
- 500 µL 1 M Tris-HCl, pH 7.5
- sterile milliQ H2O to 5 mL
Prepare fresh before each use. Adjust volume as needed based on experiment size.
Using tweezers, place filters into the lysozyme solution described above and incubate for 1 hour at 37°C.
Note: Lysozyme is an enzyme that breaks down cell wall peptidoglycan. It should be used for bacterial probes.
1h
While filters are incubating, prepare inactivation buffer (0.01 M HCl) in a 15 mL falcon tube.
0.01M HCl Inactivation Solution:
To make 10mL from 37% HCl stock, combine:
- 8.3 µL of 37% HCl
- sterile milliQ H2O up to 10 mL
Prepare fresh before each use.
After lysozme incubation, pour samples in solution onto a clean petri dish. Prepare a second clean petri dish with milliQ water. Transfer filter slices (touching only outer, cell-free edge of filter) from lysozyme solution to milliQ to wash samples. Leave filters briefly in water (5-10 seconds). Immediately place filters into 0.01M HCl inactivation buffer.
Note that most washing steps are performed most easily in petri dishes, because it is easier to handle the filters. Filters should always be handled carefully to avoid touching the part of the filter that contains the cells. It is also easier to keep filters cell side up when in petri dishes. The pencil markings on each filter slice can be used to keep track of the filter orientation.
Incubate filters in inactivation solution in 15mL tube for 10 minutes at room temperature.
10m
Wash filters in MilliQ water (in petri dish)
Wash filters in 96% Ethanol (in petri dish)
Place filters cell-side up onto whatman paper to dry.
Note: This is an optional stopping point. Filters can be prepared up to this point and stored at 4°C or -20°C. If continuing, protocol must be followed all the way through from hybridization to amplification.
Hybridization
2h 40m
Prepare hybridization buffers
Prepare and label separate 2 mL tubes with hybridization buffer for each probe (e.g. if using EUB338 and GSB532 probes, 2 separate hybridization buffer solutions should be prepared, one for each probe). Note that 300µl of hybridization buffer can hold a recommended of ~5 filter slices.
In each 2mL tube, add 300µl of hybridization buffer at the specified formamide concentration for each probe, using 50% and 0% pre-prepared hybridization stock buffers (described above) to make the desired formamide percentage. Examples below are of commonly used formamide concentrations for various probes.
- 10% formamide = 60µl of 50%stock + 240µl of 0%stock
- 20% formamide = 120µl of 50%stock + 180µl of 0%stock
- 35% formamide = 210µl of 50%stock + 90µl of 0%stock
- 40% formamide = 240µl of 50%stock + 60µl of 0%stock
Add 1 µl of desired probe working solution (50 ng DNA µl−1) to respective hybridization buffer tubes with desired formamide concentration.
Some probes also require non-HRP competitor probes (e.g. Delta495 or GAM42a), which should be described in any paper referencing these probes.
Add 1 µl of any required competitor probe working solution (50 ng DNA µl−1) to the respective hybridization buffer.
Using tweezers, place filters into the hybridization-probe mix so that the filter is facing cell-side inwards (i.e. the cell side should not be pressing against the wall of the centrifuge tube). Place filters next to each other along the wall of the tube, making sure the filter is fully submerged.
Incubate samples in hybridization buffer at 46°C for 2-3 hours.
Note that for open ocean and/or oligotrophic samples, or specific taxa known to have low ribosome numbers per cell, hybridization time may need to be extended and can continue overnight. However, in other sample types (e.g. sediments) this can cause false positives. Always be sure to include appropriate negative and positive controls and adapt the protocol to your sample type.
2h
15 minutes before the end of the incubation, warm pre-made wash buffers to 48°C (described above). Washing buffer should correspond with the % formamide of each probe (see table above) and can be made in bulk ahead of time. Aliquot washing buffers into multiple 2 mL tubes so that each filter is in excess wash buffer (3-4 filters per tube).
15m
After the hybridization incubation is complete, incubate filters in pre-warmed washing buffer for 10 minutes at 48°C.
10m
Place filters into 1X PBS and incubate at room temperature for 15 minutes. This can be done using a petri dish.
15m
During the PBS incubation, prepare amplification buffer mixture as described below.
Amplification buffer prep
Prepare 0.15% H2O2 in 1x PBS (made fresh to add to pre-made amplification buffer stock)
Combine:
5µl of 30% H2O2 in 1mL of 1x PBS or
50µl of 3% H2O2 in 1mL of 1x PBS
Using a 2mL tube, add 10µl of 0.15% H2O2 in 1xPBS and 2 µl of fluorescently labeled tyramide working solution (1 mg ml−1) (100x) to 1mL of amplification buffer (pre-made stock, described above).
For large experiments, prepare multiple 1mL amplification buffer mixtures as needed.
Note: Tyramides are very light-sensitive, keep the working solution and amplification buffer containing tyramides in the dark or covered with aluminum foil
After 15 minute PBS incubation (Step 28) is complete, dab filters/samples on blotting paper cell-side up but do not let samples dry. Proceed immediately to CARD steps below. The protocol cannot be stopped at this point.
Amplification (CARD)
40m
Place filters in 2mL tubes containing amplification buffer (with 0.15% H2O2 in 1x PBS and tyramides) and incubate at 46°C for 30 minutes in the dark.
Note: At this step, filters with different probes (i.e. from different hybridization buffers) can be combined into 2mL tubes using the same tyramide solution, as long as the same tyramide is desired.
30m
After the 30 minute incubation is complete, dab filters on blotting/whatman paper.
Incubate samples in 1X PBS for 10 minutes at room temperature in the dark. This can be done using a petri dish and covering it with aluminum foil.
10m
Wash samples briefly in MilliQ water and then 96% Ethanol. This can be done using petri dishes.
**To keep background fluorescence low, wash filters in large volumes of water and ethanol).
Let samples dry on kimwipes/whatman paper in a petri dish cell-side up. Cover petri dish with aluminum foil to keep filters in the dark.
Optional: Double Hybridization
4h 10m
If performing double hybridization (i.e. adding a second probe with second tyramide to the same sample), repeat inactivation steps by performing a 10 minute incubation in 0.01M HCl inactivation solution, wash with milliQ, followed by 96% ethanol.
Note: Can stop here and do second hybridization and amplification later.
10m
Repeat hybridization steps using second probe.
3h
Repeat amplification using a different tyramide.
1h
DAPI Counter Staining and Mounting
12m
Pipette a 10µl droplet of 1X DAPI Nucleic Acid Dye for each filter slice onto a clean petri dish. Place filter slices cell side down onto the droplet. Incubate samples for 10 minutes at room temperature in the dark (covered with Al foil).
10m
Place filter slices into a petri dish of MilliQ or 1X PBS in the dark, cell-side up. Incubate for 2 minutes at room temperature in the dark (covered with Al foil).
2m
Briefly wash filter slices in 80% ethanol to remove excess water. Place filter slices cell-side up onto a piece of Whatman paper to air dry in the dark (covered with Al foil).
Using a wide-tipped pipette, pipette ~20µL of 4:1 Citifluor:Vectashield (or other anti-fade mounting media) onto a microscope slide. Spread mixture into a line long enough for filter slices. Briefly dab filters cell-side down to coat in Citifluor:Vectashield and then rotate cell-side up and place onto the microscope slide.
Place coverslip on top of slide and gently press down with tweezers to remove any air bubbles. Wipe away the excess Citifluor:Vectashield from the edges of the slide. If desired, edges can be sealed with clear nail polish.
Freeze the slide at -20°C before visualization with an epifluorescent microscope for at least 15 minutes, but ideally for a few hours/overnight.
Solutions Overview
| Name | Molarity/Concentration | Step it's Used | Storage Conditions | |
| Lysozyme | 1000 kU/ml lysozyme, 0.05M EDTA [pH 8.0], 0.1M Tris-HCl [pH 7.5] | Permeabilization | Make Fresh | |
| HCl solution | 0.01 M | Inactivation | Make Fresh | |
| Maleic Acid Buffer | 100mM maleic acid, 150 mM NaCl (pH 7.5) | Blocking Solution Prep | Room Temperature | |
| Blocking Solution | 10% w/v Blocking Reagent | In Hybridization and Amplification Buffers | -20°C | |
| Hybridization Buffer | 0.9M NaCl, 20mM Tris-HCl [pH 7.5], 10% w/v dextran sulfate, 0.02% w/v SDS, 1% w/v blocking reagent, 0-50% formamide + probe on day of use | Hybridization | -20°C | |
| Washing Buffer | 5mM EDTA [pH 8.0], 20mM Tris-HCl [pH 7.5], 0.01% w/v SDS, 0.9-0.014M NaCl | Post-Hybridization | Room Temperature | |
| Amplification Buffer | 2M NaCl, 0.1% w/v blocking reagent, 10% w/v dextran sulfate, 1xPBS + H2O2 and tyramide on date of use | Amplification | -20°C |
Summary of solutions used in protocol and their corresponding step and storage temperature.
Cell Enumeration
From sample prep, note the inner radius of the filter tower used to filter samples and determine the area of the filter that was in contact with the sample in µm^2 (A).
Note the sample dilution factor (D) and volume of sample filtered in µl (V).
On the microscope, note grid area in µm^2 of the the magnification used for counting (G).
Count the number of cells in the grid. Repeat over 20 random areas on a given filter slice.
Convert each cell count number (C) to cell concentration by the following:
Cell concentration (cells/mL) = C (A/G) * (D/V) * 1000
Cell concentration (cells/mL) = number of cells in the grid * (area of the filter/area of the grid) * (dilution factor/volume filtered) * 1000
If samples used in CARD-FISH were centrifuged during preparation, count DAPI stained cells on CARD-FISH samples as well as on non-centrifuged samples that remained in PFA and were not used in CARD-FISH.
Compare these two values and generate a correction factor that represents the % loss due to centrifugation, and then multiply the CARD-FISH cell counts by this correction factor.
Protocol references
Pernthaler A, Pernthaler J, Amann R.2002.Fluorescence In Situ Hybridization and Catalyzed Reporter Deposition for the Identification of Marine Bacteria. Appl Environ Microbiol68:.https://doi.org/10.1128/AEM.68.6.3094-3101.2002
Ishii, K, Mußmann, M., MacGregor, B.J., Amann, R. 2004. An improved fluorescence in situ hybridization protocol for the identification of bacteria and archaea in marine sediments, FEMS Microbiology Ecology, Volume 50, Issue 3, Pages 203–213, https://doi.org/10.1016/j.femsec.2004.06.015
Herndel, Gerhard et al. 2018. CARD-FISH and Microautoradiography Protocol for Bacteria and Archaea.
Teira E, Reinthaler T, Pernthaler A, Pernthaler J, Herndl GJ. Combining catalyzed reporter deposition-fluorescence in situ hybridization and microautoradiography to detect substrate utilization by bacteria and Archaea in the deep ocean. 2004. Appl Environ Microbiol. 70(7):4411-4. doi: 10.1128/AEM.70.7.4411-4414.2004. PMID: 15240332; PMCID: PMC444763. https://pubmed.ncbi.nlm.nih.gov/15240332/
Ruff SE, Arnds J, Knittel K, Amann R, Wegener G, Ramette A, Boetius A. 2013. Microbial communities of deep-sea methane seeps at Hikurangi continental margin (New Zealand). PLoS One. Sep 30;8(9):e72627. doi: 10.1371/journal.pone.0072627. PMID: 24098632; PMCID: PMC3787109. https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0072627
Acknowledgements
We thank Hannah Vanderscheuren, Anna Warsaw, and Daniela Pierro for their feedback in the lab and on the written version of this protocol.