Jan 24, 2025
Building Synthetic Long Non-Coding RNAs via Iterative Golden Gate Assembly of K-mer Arrays
- Seong Hu Kim1,
- Chloe Guerrero2,
- Karmella A Haynes1
- 1Wallace H. Coulter Department of Biomedical Engineering, Emory University, Atlanta, GA, USA;
- 2Department of Biology, Emory University, Atlanta, GA, USA
- Karmella A Haynes: Corresponding author;
- Haynes Lab at Emory



Protocol Citation: Seong Hu Kim, Chloe Guerrero, Karmella A Haynes 2025. Building Synthetic Long Non-Coding RNAs via Iterative Golden Gate Assembly of K-mer Arrays. protocols.io https://dx.doi.org/10.17504/protocols.io.8epv52bxjv1b/v1
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: January 03, 2025
Last Modified: January 24, 2025
Protocol Integer ID: 117653
Keywords: Long noncoding RNA, synthetic biology, Golden Gate assembly, synthetic lncrna, repeat motifs within lncrna, understanding lncrna mechanism, noncoding rna, iterative golden gate assembly, rna construct, lncrna element, rna, lncrna, golden gate assembly, gene transcription, chromatin organization, nucleotide linker, chromatin complex recruitment, systematic investigation of repeat motif, bbsi golden gate cloning site, rna polymerase ii, repeat motif, nucleotide, building synthetic long non, molecular guide, mer arrays long, gene
Funders Acknowledgements:
National Science Foundation
Grant ID: 2243665
Disclaimer
The authors declare no conflicts of interest.
Abstract
Long noncoding RNAs (lncRNAs), a class of noncoding RNAs exceeding 500 nucleotides and transcribed mostly by RNA polymerase II, regulate gene transcription and chromatin organization. Acting as molecular guides, scaffolds, and structural components, lncRNAs interact with RNAs, DNA, and proteins. Repeat motifs within lncRNAs called k-mers are associated with protein interactions and chromatin complex recruitment. Understanding lncRNA mechanisms is hampered by tissue specificity, redundancy, and multifunctionality, necessitating quantitative investigation. To support the systematic investigation of repeat motifs, we optimized Golden Gate assembly with standardized 4-nucleotide linkers to assemble any four lncRNA elements into 4x k-mers without the need to design compatible overhangs. To extend the length (repeat number) of RNA constructs, a 2x BbsI Golden Gate cloning site is restored at the 3’ end of the assembled 4x k-mer, allowing the addition of more k-mers to generate an 8x k-mer, 12x k-mer, and so on. Please read the Guidelines for suggestions on how to make arrays of intermediate lengths (e.g. 1x, 2x, 3x, 5x, 6x, 7x, etc.). Beginning with k-mer identification, synthetic lncRNAs can be constructed and expressed in vitro or in cells within three weeks, offering an efficient framework to study and harness their regulatory functions.
Image Attribution
All graphics were generated in BioRender.
Guidelines
Introduction
Long non-coding RNAs (lncRNAs) are RNA transcripts over 500 nucleotides, primarily synthesized by RNA polymerase II (Pol II) [1]. LncRNAs contain repeat motifs that have been associated with biological activities including protein scaffolding, chromatin localization, and regulation of mRNA stability, translation, and decay. Similar to protein domains, lncRNA repeat motifs are arranged linearly within the longer primary sequence, and their spatial arrangement supports proper function of the entire macromolecule [2]. Although lncRNAs are usually expressed at much lower levels than messenger RNAs [3], many have a surprisingly large impact on cellular biology and molecular processes. For instance, the lncRNA Xist (19,000 nucleotides) initiates epigenetic silencing of one entire X chromosome in XX female mammals, yet XIST lncRNAs are found in just around 50 “hubs” along the inactive X-chromosome (Xi) at steady-state silencing (maintenance), totalling just 50 - 100 molecules per Xi [4]. Nevertheless, XIST still controls the silencing of around 85% of about 800 genes across the 170 Mb Xi [5,6]. Although mechanisms like phase separation and target-directed miRNA degradation have been suggested to explain the sub-stoichiometric effects of lncRNAs [7], our ability to fully understand their extensive roles is constrained by the lack of theory-driven, quantitative investigations. Moreover, despite the insights gained from reverse-genetics approaches, the tissue-specific expression, redundancy, and diverse functional profiles of lncRNAs often confound phenotype-activity relationships, underscoring the need for more rigorously controlled strategies [8].
LncRNAs other than XIST also shape gene expression by orchestrating multiple layers of epigenetic regulation. They can restructure the chromatin landscape, as demonstrated by the capacity of LncMyoD to modify chromatin accessibility and direct muscle stem cell differentiation [9]. LncRNAs also form functional partnerships with epigenetic complexes, as illustrated by SWINGN’s interaction with the SWI/SNF complex to enhance oncogene promoter accessibility, and NEAT1’s binding to BRD4 and WDR5 to interfere histone modifications and transcriptional activity [10,11]. LncRNAs can establish persistent epigenetic states, as evidenced by Firre’s ability to maintain stable transcriptional changes over time [12]. Notably, upon its induction, Firre rapidly modulates epigenetic and transcriptional programs in trans, exerting these effects within as little as 30 minutes. Collectively, these mechanisms demonstrate the dynamic and sustained regulatory capabilities of lncRNAs in governing epigenetic and transcriptional landscapes.
Minimal Functional Units of lncRNAs: Stem-loops, Triplexes, and K-mers
A stem-loop is a secondary structural conformation composed of a Watson-Crick base paired stem and a long single stranded loop. Similar structures, referred to as hairpins, have shorter single stranded loops. Several studies have taken structures from computational, cryo-electron microscopy, chemical probing studies [13] and altered their sequences to manipulate folding and stability to determine the contribution of stem-loops to the biological activity of lncRNAs in cells. For instance specific stem-loop structures, such as AUCG hairpins in XIST, significantly affect their ability to recruit epigenetic modifiers, such as histone methyltransferases, thereby altering gene expression patterns [14]. Mutations introduced into tandem stem-loops of the Drosophila roX2 lncRNA disrupted its functionality in male X-chromosome dosage compensation [15].
Triplexes are structures where three single-stranded nucleic acids, including RNA or both RNA and DNA, form a helix. In DNA-RNA triplex structures, RNA binds to the purine-rich strand of DNA via Hoogsteen or reverse Hoogsteen hydrogen bonds, anchoring RNA to specific sequences to enable targeted regulation of gene expression at local and distant genomic sites [16]. Computational tools such as Triplex Domain Finder (TDF) have identified lncRNAs with triplex-forming potential, such as GATA6-AS, which regulate gene expression during cardiac differentiation [17]. LncRNA PAPAS has been shown to regulate rRNA gene transcription under stress by forming a DNA-RNA triplex structure that recruits the CHD4/NuRD complex to enhancers, facilitating chromatin remodeling and transcriptional repression [18]. In addition to DNA-RNA triplex structures, there are bipartite triplexes composed entirely of RNA. This structural motif contains two distinct regions of triple-helix stacking interrupted by specific nucleotides that help maintain alignment and stability. An example is the lncRNA MALAT1, which contains five and four U-A-U triple helices separated by a C-G-C triplet and a C-G doublet, stabilized further by interactions involving the insertion of adenine bases into the minor groove of adjacent helices, effectively preventing rapid RNA decay [19].
The study of k-mers, short nucleotide sequences of “k” length, has significantly advanced the analysis and understanding of lncRNAs. K-mers are instrumental in deciphering the functional and structural characteristics of lncRNAs, particularly given their frequent lack of linear sequence homology. It has been demonstrated that lncRNAs with similar k-mer profiles often share functional similarities, such as protein-binding affinities and subcellular localization patterns, thereby providing insights into their regulatory mechanisms [20]. Moreover, k-mers are pivotal in predicting the subcellular localization of lncRNAs through machine learning models like lncLocPred, which leverage k-mer features to achieve high prediction accuracy [21]. Beyond classification and localization, k-mers also influence the stability and interaction dynamics of lncRNAs. They mediate critical protein-RNA and RNA-RNA interactions that determine the stability of lncRNAs, affecting their degradation rates and involvement in regulatory networks [22]. Collectively, k-mer analysis overcomes the complexity of lncRNA sequences, enhancing our ability to classify, predict, and understand their diverse functions.
Exploring the Functional Modularity of lncRNA Sequences
Synthetic biology can be used to validate or refine insights into the function of the minimal structural units of lncRNAs. Current insights have come from using traditional approaches, such as genetic deletion [23], ChIRP, Shape-seq, CLIP-seq, FISH, CRISPR-based screens, to investigate natural lncRNAs. A large body of work has generated a structural model for XIST (Fig 1A). Predicted lncRNA monomers derived from lncRNAs including XIST and others can be used as modules to assemble synthetic functional RNAs. Comparing tunable parameters such as low to high k-mer repeat number [24], alternative base-pairing within predicted stem-loops [24], alternative nucleotides in single-stranded loops [25], and low to high computationally predicted protein binding scores [26].
Engineered lncRNA systems have successfully modulated gene expression and protein translation across various contexts. Ectopic expression of synthetic lncRNAs near a natural target gene or reporter gene can be used to measure epigenetic regulation (Fig 1B). For instance, distinct regions of human XIST responsible for silencing and localization were identified [27], and assembled into synthetic RNAs to demonstrate the additive silencing effect of A-repeat k-mers [24]. Additionally, a minimal XIST transgene incorporating the Polycomb Interaction Domain from mouse Xist was developed, enhancing gene silencing and heterochromatin recruitment [28]. The RNA enhancement (RNAe) system utilizes an artificial lncRNA containing a SINEB2 repeat from a lncRNA AS-UCHL1 to enhance targeted protein translation without affecting mRNA levels [29]. Building upon this, the CRISPR/dCasRx-SINEB2 system demonstrated a significant reduction in off-target effects while effectively recruiting RNA-binding proteins to target mRNAs, making it a precise tool for translational control [30]. Finally, CRISPR-Display incorporated functional lncRNA domains directly into the sgRNA scaffold. This allowed precise genomic targeting while retaining the regulatory capabilities of these lncRNAs at target loci, enabling multiplexed functional studies [31]. Results generated from synthetic systems can potentially support future rational design of synthetic RNAs with desired behaviors for cellular epigenetic engineering.
Figure 1 | Approach: LncRNA structure-guided experimental design using XIST as an example. (A) Annotated map of the XIST lncRNA [32]. The map highlights regions A - F, which have been defined through comparative evolutionary biology, genetic dissection, protein binding analysis, RNA structure analyses, or a combination of approaches. Repeat region A contains 9 repeat motifs (k-mers) of slightly different lengths (k) [24], shown here mapped onto transcript NCBI M97168. (B) Schematic of how synthetic biology and quantitative analysis can be used to refine understanding of the biological function of epigenetic-silencer lncRNAs such as XIST. NC = negative control (no synthetic lncRNA).
Experimental Design
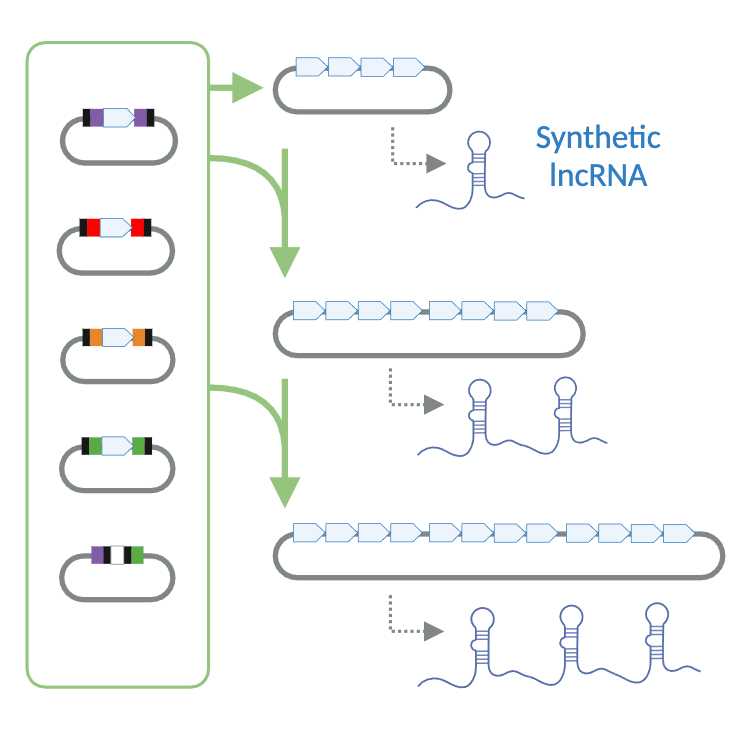
The workflow for assembling synthetic lncRNAs in an intermediate cloning vector and then into the expression vectors described herein can be completed in about 9, 14, or 19 days for 4x, 8x, and 12x k-mer constructs, respectively (Fig. 2). The workflow begins with synthesizing donor k-mers as double-stranded DNA, which are then blunt-ligated into kanamycin-resistant (TOPO‱) vectors. These vectors are transformed into competent cells, and the resulting colonies are screened using colony PCR. This stage typically takes around three days. The verified donor constructs are subsequently used in single-step Golden Gate assembly reactions to generate 4x, 8x, and 12x k-mer arrays through iterative assembly and reintroduction of BbsI cloning sites. Each assembly phase, including validation through gel electrophoresis and sequencing, generally requires three days. The finalized constructs can then be inserted into mammalian expression systems for functional studies, enabling detailed investigations into lncRNA regulatory functions and molecular interactions.
Figure 2 | Timeline and overview of steps.
K-mer Donor Module Design for Golden Gate Assembly
The Golden Gate method for lncRNA k-mer assembly has important advantages over other methods such as scarless Gibson assembly and de novo DNA synthesis. In regard to Gibson assembly, the very short length of lncRNA k-mers (~50 nt) renders them vulnerable to complete destruction during the exonuclease step that is used to create sticky single-stranded overhangs. Our approach uses an endonuclease to generate 4 nt overhangs between k-mers. One important advantage of our Golden Gate linkers is that these allow the reuse of the same compatible sticky ends, speeding up the assembly process by obviating the need to redesign optimal 4 bp overhangs for every assembly. In regard to de novo synthesis, highly repetitive sequences like those found in lncRNAs are a challenge because they can lead to secondary structure formation, synthesis errors, and misannealing during oligonucleotide assembly [33].
As described in our previous protocol for building fusion protein-encoding open reading frames, single-pot Golden Gate assembly uses two essential types of DNA fragments, donor and destination, in a one-step digestion-ligation reaction [34]. In our current protocol, each donor module (A, B, C, or D) contains a non-coding lncRNA k-mer or other sequence of interest flanked by unique 4-bp spacers and BbsI recognition sites (Fig. 3A). Donor plasmids are generated through Zero Blunt‱ TOPO‱ cloning into PCR-Blunt II-TOPO and constructs are verified by colony PCR and Sanger sequencing. PCR amplification to generate linear donors is not recommended because lncRNA sequences are often low-complexity and repetitive, making it difficult to design highly specific primers. Upon digestion with BbsI, these recognition sites are removed, leaving 4-nt 5′ overhangs at each end that dictate the sequential order of the modules (A - B - C - D). The 4 bp “scars” produced after assembly might be of concern, since lncRNA structure and function is directly linked to primary structure (Fig. 3A). However, Minks et al [24] successfully built a functional XIST-derived 9x 46-mer that contained restriction enzyme cut sites between A-repeat k-mers.
Figure 3 | Detailed schematic of key steps from Figure 1. (A) Assembly of a 4x K-mer Array in the GGDest5-Amp Destination Vector. The first and last base pairs (nn) of each k-mer module are positioned next to a 4-bp site that, after cleavage by BbsI, guides directed assembly of the modules (A, B, C, and D). Single-pot digestion and ligation with Type IIS enzyme BbsI and T4 DNA ligase results in four modules linked by 4 bp scars. (B) Detailed map of the GGDest5-Amp destination vector.
Assembly of 4x, 8x, and 12x K-mer Arrays
To construct progressively larger arrays (4x, 8x, or 12x), the same Golden Gate workflow is repeated in successive rounds. First, a 4x construct is generated by assembling four donor modules into the GGDest5-Amp backbone [35]. Next, the BbsI cloning site is reestablished in this 4x plasmid (Fig. 4), enabling the addition of four new k-mer modules in a single-pot Golden Gate reaction to yield an 8x array. This restoration step also allows the 8x construct to serve as the destination vector for assembling 12x arrays. Each iterative cycle consists of (i) preparing the destination vector and donor modules, (ii) performing the digestion-ligation reaction under optimized thermocycler conditions, (iii) transforming the reaction product and screening resulting colonies by colony PCR, and (iv) purifying and verifying recombinant DNA by restriction analysis and Sanger sequencing (Fig. 2). This streamlined “digest, ligate, restore” strategy can be repeated as needed, using the plasmid from one assembly round as the substrate for the next round.
Figure 4 | Restoration of the BbsI Golden Gate Cloning Site After the 4x K-mer. The Golden Gate cloning site (2x BbsI) is regenerated by inserting a short double stranded DNA oligo (dsOligo) into a BcuI/ PstI site downstream of the k-mer array. The BcuI site on the 5’ side of 2x BbsI is destroyed by introducing a T/A base pair (*), and a new BcuI site is introduced on the 3’ side to allow subsequent drop-ins.
Assembling Arrays of Intermediate Sizes
Arrays can be reduced from four to three, two, or one k-mer by generating “hybrid” modules that start and end with different overhangs. For instance, a 1x k-mer can be built in GGDest5-Amp by using a single A-D hybrid module, where the k-mer sequence is flanked with A-left (5’) and D-right (3’) overhangs. A 2x k-mer can be built by assembling an A k-mer module with another one flanked by B-left and D-right overhangs. To build a 3x k-mer, an A k-mer and B k-mer can be assembled with a third k-mer flanked by C-left and D-right overhangs.
Longer arrays, such as 5x, 6x, and 7x k-mers can be built from a pre-existing 4x k-mer construct. After the 2xBbsI cloning site is regenerated downstream of the 4x array, a 5x array can be built by inserting a single A-D hybrid module. A 6x array can be built by inserting an A k-mer module plus one flanked by B-left and D-right overhangs. Finally, a 7x array can be built using an A k-mer, a B k-mer, and a k-mer flanked by C-left and D-right overhangs. A similar strategy can be used to build 9x, 10x, and 11x arrays after the 2xBbsI site is generated downstream of an 8x k-mer array. This step-wise approach allows tightly controlled and reliable assembly of arrays of n-length.
Transferring Synthetic K-mer Constructs to Expression Vectors
To determine the biological activity of synthetic lncRNAs in cells, the array must be cloned downstream of a promoter in an expression vector. Here, we provide recommendations for two systems with drug-inducible promoters for tight regulation of synthetic lncRNA expression levels. First, pcDNA5 FRT TOPO (Thermo Fisher #V652020) is an inducible expression vector designed for tetracycline-regulated gene expression in mammalian cells. Integration requires a host cell with a single FRT landing pad site and Flp recombinase for site-specific recombination. Host cells must stably express the Tet repressor for CMV/TetO2 promoter regulation and be hygromycin-sensitive for transfectant selection. A k-mer array can be inserted downstream of the promoter by ligating a NotI digested array with a NotI digested and dephosphorylated pcDNA5 FRT TOPO plasmid. Since ligation is non-directional, it is important to verify forward inserts via Sanger or Nanopore sequencing. Second, to facilitate introduction of a synthetic lncRNA-expressing gene semi-randomly across the genome, we generated pSBtetTA-YP_NotISpeI which is a modified version of the pSBtet-GP plasmid [36]. Like pcDNA FRT TOPO, this vector contains a dox-inducible CMV-derived promoter. However, it does not require a “landing pad” in the host cell, and the vector expresses the Tet regulator protein so no additional transgene is required to regulate the promoter. We introduced NotI and SpeI downstream of the promoter to facilitate cloning. We also replaced GFP with YFP Venus to avoid spectral overlap with other fluorophores for potential downstream multi-color experiments. This plasmid is available from our lab and a fully annotated sequence is available at Benchling [37].
To study the activity of an lncRNA using the pcDNA5 FRT TOPO system, a stable cell line expressing the lncRNA of interest can be created through co-transfection with the pOG44 plasmid (Thermo Fisher #V600520), which provides Flp recombinase activity for site-specific integration at the FRT landing pad site within the host genome. GFP silencing experiments can be conducted to evaluate the lncRNA's functional effects, following methods previously shown to silence flanking reporter genes [24]. GFP expression can be measured using fluorescence microscopy to assess the silencing efficiency. Additionally, RT-qPCR can be used to analyze the expression of GFP and endogenous genes near the FRT integration site, such as CLDN16 and IL1RAP, which were identified as silencing targets of the lncRNA [24].
The pSBtetTA-YP_NotISpeI system provides a flexible approach to studying lncRNA activity by enabling the generation of stable cell lines through co-transfection with the plasmid and the pCMV(CAT)T7-SB100 transposase expression vector. The SB100X transposase facilitates semi-random genomic integration of the plasmid into the host cell’s genome [38]. Functional studies can be performed by conducting pRPBSA-YFP gene silencing experiments, where YFP expression levels are measured using fluorescence microscopy to assess lncRNA-mediated regulatory effects. To determine the site of genomic integration, the genomic sequence next to either inverted terminal repeat (ITR) can be isolated and sequenced using techniques such as Splinkerette PCR [38]. This mapping enables researchers to connect the observed effects of the lncRNA to specific genomic loci. RT-qPCR can be utilized to evaluate changes in the expression of nearby endogenous genes, offering insights into the lncRNA’s potential influence on local chromatin organization and gene regulation.
Materials
REAGENTS
Single Stranded DNA Oligos (without 5’ phosphates)
M13F, 5’-GTAAAACGACGGCCAG
M13R, 5’-CAGGAAACAGCTATGAC
Top 2xBbsI Oligo, 5’-CTAGAGGAGGGGTCTTCGAGAAGACCTACTAGTAGCGGCCGCTGCA
Bottom 2xBbsI Oligo, 5’-GCGGCCGCTACTAGTAGGTCTTCTCGAAGACCCCTCCT
CMV-Forward, 5’-CGCAAATGGGCGGTAGGCGTG
BGHR, 5’-TAGAAGGCACAGTCGAGG
pSBtetTA-YP_F, 5’-ATCGCCTGGAGCCAATTCC
pSBtetTA-YP_R, 5’-AACCTCCCACATCTCCCCC
K-mer Donor Plasmid preparation
Duplex oligos of lncRNA derived modules (k-mers) for Golden Gate assembly (see Table 1)
Nuclease-Free Water, for Molecular BiologyMerck MilliporeSigma (Sigma-Aldrich)Catalog #W4502-1L
Zero Blunt™ TOPO™ PCR Cloning Kit without competent cellsThermo Fisher ScientificCatalog #450245
Golden Gate Assembly Reaction
GGDest5-Amp, Haynes Lab
DNA modules A, B, C, D in Zero Blunt‱ TOPO‱ plasmid
T4 DNA Ligase Reaction BufferNew England BiolabsCatalog #B0202S
Quick Ligation Kit - 150 reactionsNew England BiolabsCatalog #M2200L
FastDigest BpiI (BbsI) (IIs class)Thermo ScientificCatalog #FD1014
Nuclease-Free Water, for Molecular BiologyMerck MilliporeSigma (Sigma-Aldrich)Catalog #W4502-1L
2x BbsI dsOligos Preparation
Top 2xBbsI Oligo, 5’-CTAGAGGAGGGGTCTTCGAGAAGACCTACTAGTAGCGGCCGCTGCA
Bottom 2xBbsI Oligo, , 5’-GCGGCCGCTACTAGTAGGTCTTCTCGAAGACCCCTCCT
T4 DNA Ligase Reaction BufferNew England BiolabsCatalog #B0202S
T4 Polynucleotide KinaseNew England BiolabsCatalog #M0201S
Nuclease-Free Water, for Molecular BiologyMerck MilliporeSigma (Sigma-Aldrich)Catalog #W4502-1L
DNA Dephosphorylation, Deactivation, Purification, & Ligation
4x or 8x k-mer GGDest5-Amp plasmid
2x BbsI dsOligo
FastDigest Buffer (10X)Thermo Fisher ScientificCatalog #B64
FastDigest BcuI (SpeI)Thermo Fisher ScientificCatalog #FD1254
FastDigest PstIThermo FisherCatalog #FD0614
Quick CIPNew England BiolabsCatalog #M0525S
Nuclease-Free Water, for Molecular BiologyMerck MilliporeSigma (Sigma-Aldrich)Catalog #W4502-1L
DNA Clean & Concentrator-25 (Capped)Zymo ResearchCatalog #D4033
T4 DNA Ligase Reaction BufferNew England BiolabsCatalog #B0202S
Quick Ligation Kit - 150 reactionsNew England BiolabsCatalog #M2200L
K-mer Array Transfer from Destination Vectors to Expression Vectors
4x, 8x, or 12x k-mer GGDest5-Amp plasmid
pSBtetTA-YP_NotISpeI, Haynes Lab
pcDNA™5/FRT/TO Vector KitInvitrogenCatalog #V652020
FastDigest NotIThermo FisherCatalog #FD0594
FastDigest BcuI (SpeI)Thermo Fisher ScientificCatalog #FD1254
Quick CIPNew England BiolabsCatalog #M0525S
FastDigest Buffer (10X)Thermo Fisher ScientificCatalog #B64
Nuclease-Free Water, for Molecular BiologyMerck MilliporeSigma (Sigma-Aldrich)Catalog #W4502-1L
T4 DNA Ligase Reaction BufferNew England BiolabsCatalog #B0202S
Quick Ligation Kit - 150 reactionsNew England BiolabsCatalog #M2200L
FastDigest EcoRIThermo Fisher ScientificCatalog #FD0274
FastDigest Green Buffer (10X)Thermo Fisher ScientificCatalog #B72
Colony PCR
Nuclease-Free Water, for Molecular BiologyMerck MilliporeSigma (Sigma-Aldrich)Catalog #W4502-1L
DreamTaq Green PCR Master Mix (2X)Thermo FisherCatalog #K1082
Bacterial Transformation & Liquid Bacterial Culture
Quick Ligation Kit - 150 reactionsNew England BiolabsCatalog #M2200L
T4 DNA Ligase Reaction BufferNew England BiolabsCatalog #B0202S
Nuclease-Free Water, for Molecular BiologyMerck MilliporeSigma (Sigma-Aldrich)Catalog #W4502-1L
DNA Clean & Concentrator-25 (Capped)Zymo ResearchCatalog #D4033
NEB Turbo Competent E.coli (High Efficiency) - 20x0.05 mlNew England BiolabsCatalog #C2984H
SOC Outgrowth Medium - 100 mlNew England BiolabsCatalog #B9020S
LB Broth (Lennox)Merck MilliporeSigma (Sigma-Aldrich)Catalog #L3022
Sodium hydroxideMerck MilliporeSigma (Sigma-Aldrich)Catalog #795429-500G
LB Broth with agar (Lennox)Merck MilliporeSigma (Sigma-Aldrich)Catalog #L7533
Ampicillin sodium saltMerck MilliporeSigma (Sigma-Aldrich)Catalog #A9518
Kanamycin sulfateMerck MilliporeSigma (Sigma-Aldrich)Catalog #60615
Plasmid Purification and Verification
ZR Plasmid Miniprep - ClassicZymo ResearchCatalog #D4016
FastDigest EcoRIThermo Fisher ScientificCatalog #FD0274
FastDigest BcuI (SpeI)Thermo Fisher ScientificCatalog #FD1254
FastDigest BpiI (BbsI) (IIs class)Thermo ScientificCatalog #FD1014
FastDigest Green Buffer (10X)Thermo Fisher ScientificCatalog #B72
Nuclease-Free Water, for Molecular BiologyMerck MilliporeSigma (Sigma-Aldrich)Catalog #W4502-1L
Gel Electrophoresis
SYBR SAFE DNA stainInvitrogen - Thermo FisherCatalog #S33102
GeneRuler 1 kb DNA Ladder, ready-to-useThermo FisherCatalog #SM0313
Agarose, Low EEO/MultipurposeFisher ScientificCatalog #BP160-100
50X Tris Acetate-EDTA buffer (TAE) Electrophoresis BufferThermo ScientificCatalog #B49
CONSUMABLES
Tips for Rainin LTS 20 µLpipette.comCatalog #LE-20
200 uL LTS Compatible Unfiltered Low Retention Pipette TipsOxford Lab ProductsCatalog #LTR-200-LR
Tips for Rainin LTS 1000 µL (1200 µL max)pipette.comCatalog #LE-1000
CAPP Harmony Serological Pipette Variety Pack (5mL 10mL and 25mL)pipette.comCatalog #SP-VAR1
Microcentrifuge Tubes: 0.6 mLFisher ScientificCatalog #05-408-120
1.5 mL microcentrifuge tubesFisher ScientificCatalog #05-408-129
Microcentrifuge Tubes: 2.0 mLFisher ScientificCatalog #05-408-138
MicroAmp™ 8-Tube Strip with Attached Domed Caps 0.2 mLThermo Fisher ScientificCatalog #A30589
15 mL Centrifuge Tube with Flat CapOxford Lab ProductsCatalog #OCT-15B
PR1MA™ 50 mL Conical Centrifuge TubesMIDSCICatalog #C50B
500 mL Round Media Storage Bottles, with GL45 Screw CapCorningCatalog #1395-500
1L Round Media Storage Bottles, with GL45 Screw CapCorningCatalog #1395-1L
1L Reusable Plastic Graduated CylinderCorningCatalog #3022P-1L
15mm Polystyrene Petri DishesCorningCatalog #07-202-010
MilliporeSigma™ Novagen™ ColiRollers™ Plating BeadsFisher ScientificCatalog #71-013-4
PYREX® Erlenmeyer Flasks Graduated Wide Mouth (250 mL)VWR International (Avantor)Catalog #29140-045
Nunc™ 14 mL Round-Bottom TubeFisher ScientificCatalog #12-565-971
Bemis™ Parafilm™ M Laboratory Wrapping FilmFisher ScientificCatalog #13-374-12
Ice / Dry Ice Bucket (EVA Foam)Fisher ScientificCatalog #03-395-152
EQUIPMENT
Equipment
ProFlex 3x32 well PCR System
NAME
PCR system
TYPE
Thermo Fisher
BRAND
44-840-73
SKU
LINK
Equipment
Shaking Incubator - MaxQ 4450
NAME
Incubator Shaker
TYPE
Thermo Scientific
BRAND
SHKE4450
SKU
LINK
Equipment
iBright FL1000 gel and blot imager
NAME
Imaging System
TYPE
Thermo Fisher
BRAND
A32752
SKU
LINK
Discontinued
SPECIFICATIONS
Equipment
Compact Dry Baths/Block Heaters, Model D
NAME
Dry Bath
TYPE
Thermo Scientific
BRAND
10753-608
SKU
LINK
Equipment
Owl™ EasyCast™ B1 Mini Gel Electrophoresis Systems
NAME
Gel electrophoresis apparatus
TYPE
Thermo Scientific
BRAND
B1-BP
SKU
LINK
Equipment
Nanodrop One
NAME
UV-Vis Spectrophotometer
TYPE
Thermo Scientific
BRAND
13-400-518PRM
SKU
LINK
Equipment
Microfuge 16 Centrifuge, Non-Refrigerated
NAME
Centrifuge
TYPE
Beckman Coulter
BRAND
A46474
SKU
LINK
Equipment
BenchMixer™ Vortexer mixer
NAME
Vortexer
TYPE
Benchmark Scientific
BRAND
BV1000
SKU
LINK
Equipment
Heratherm™ General Protocol Microbiological Incubators
NAME
Incubator
TYPE
Thermo Scientific
BRAND
51028063
SKU
LINK
Equipment
Countertop Microwave
NAME
Microwave
TYPE
Panasonic
BRAND
NN-SD765S
SKU
LINK
Equipment
PowerPac™ Basic Power Supply
NAME
Power supply
TYPE
BIO-RAD
BRAND
1645050
SKU
LINK
Equipment
P2-Series Balances
NAME
Balance
TYPE
VWR
BRAND
VWR-203P2
SKU
LINK
REAGENT SETUP
TAE electrophoresis solution: Dilute TAE buffer solution in distilled water to 1X, and keep at room temperature for up to 6 months.
LB liquid growth media: Prepare 1 liter of LB liquid media by dissolving 20 g of Lennox LB Broth granules (or equivalent pre-mixed formulation) in ~500 mL deionized water. Add 1 mL of 1M NaOH, then bring the solution to a final volume of 1 liter with deionized water. Distribute 200 mL portions into sterilized bottles, loosely cap them, and autoclave for 15 minutes on a liquid sterilization cycle. Once cooled, tighten the caps, label the bottles, and store at room temperature or 4°C (avoid freezing). Add appropriate antibiotics.
Agar plates: Dissolve 17.5 g LB broth with agar in 500 mL distilled water and autoclave on a short liquid cycle (00:20:00 sterilization, no drying) to fully melt the agar. Let the buffer cool to 60 °C before adding the appropriate antibiotic. Pour 10 mL of LB broth with agar plus antibiotic per plate and let the agar cool overnight before storing the plates at 4 °C stacked and inverted (lid-side-down).
Troubleshooting
Safety warnings
Personal Protective Equipment (PPE): At minimum, a standard cotton/ polyester lab coat and disposable nitrile (or similar) gloves. Biohazard: Treat all cell culture liquid waste with bleach (10% final concentration) and dispose of the treated waste according to your institute’s environmental health and safety (EH&S) protocol. Discard all disposable items (e.g. micropipette tips, culture tubes, etc.) that have come into contact with growth medium and/or cells as dry biohazard waste. Recombinant DNA: The plasmid vectors described in this protocol are not known to support transmission between cells or organisms or high rates of horizontal gene transfer in the environment. Chemical Warning: Do not mix ethanol waste with bleach waste. Doing so will produce toxic chloroform vapors.
Note: This text comes directly from our previous molecular cloning protocol “Rapid Single-Pot Assembly of Modular Chromatin Proteins for Epigenetic Engineering V.1” (2021) dx.doi.org/10.17504/protocols.io.brgcm3sw
Generation of Donor K-mers
19h 8m 45s
Design Golden Gate-compatible k-mers. Review the double stranded DNA (dsDNA) synthesis specifications for your preferred vendor. We recommend using Integrated DNA Technology’s (IDT’s) Duplexed DNA service, with a size range of 10 - 90 bp (100 nmole Duplex Oligo). Design monomer sequences as described in the Table 1 below for Duplexed DNA. If your sequences of interest are longer than 90 bp you may consider Duplexed Ultramers (45 - 200 bp), or IDT gBlocks (125 - 3000 bp). Have the dsDNA synthesized without 5’ phosphates.
| A | B | C | D | |
| BbsI Left (5'-3') 14 bp | LncRNA k-mer sequence | BbsI Right (5'-3') 14 bp | ||
| Module A | TTGAAGACCTGGAG | Up to 62 nt | TCTGGGGTCTTCAA | |
| Module B | TTGAAGACCTTCTG | Up to 62 nt | CCATGGGTCTTCAA | |
| Module C | TTGAAGACCTCCAT | Up to 62 nt | GCCCGGGTCTTCAA | |
| Module D | TTGAAGACCTGCCC | Up to 62 nt | ACTAGGGTCTTCAA |
TABLE 1 | Design of lncRNA derived modules (k-mers) for Golden Gate assembly.
Resuspend the dsDNA. Bring the DNA fragments to 100 µM with MB H2O. Dissolve with gentle heating (50°C) and occasional vortexing for 10 minutes.
15m
Ligation of k-mers into plasmids through Zero Blunt‱ TOPO‱ PCR Cloning. The dsOligo is subjected to a Zero Blunt‱ TOPO‱ PCR Cloning reaction in a PCR tube according to the reaction table below. Reaction time is 10 minutes at room temperature.
| A | B | C | |
| Reagent | Volume (µL) | Final Concentration | |
| dsDNA (100 µM) | 0.5 | 10 µM | |
| *Salt solution | 1.0 | ||
| *pCR-Blunt II-TOPO | 1.0 | ||
| MB H2O | 3.5 |
*Reagents provided in the Thermo Fisher Scientific Zero Blunt‱ TOPO‱ PCR Cloning Kit.
15m
Transformation of ligated products.
Pipette the 6 µL reaction product into a 2.0 mL microcentrifuge tube and add 50 µL chemically competent DH5ɑ-T cells. Gently tap the tube three times and incubate on ice for 5 minutes.
5m
Heat shock at 42°C for 45 seconds.
45s
Add 1 mL SOC outgrowth medium and incubate on a shaking platform at 320 rpm, 37°C for 30 minutes.
30m
Pellet the cells at 16,300 x g, room temperature, for 3 minutes and discard the supernatant.
3m
Resuspend the pellet(s) in 50 µL 50 µg/mL kanamycin LB broth. Plate each resuspension onto a pre-warmed 50 µg/mL kanamycin LB agar plate and spread using beads. Incubate at 37°C overnight.
18h
Recombinant Plasmid Screening with Colony PCR
2h 27m
Plate inspection. Obtain colony plates from the incubator and inspect for colony growth. Optimal growth for colony PCR will have clearly separated colonies with minimal to no surrounding satellites, allowing precise colony picking with no cross-contamination. Determine the number of colonies to be picked based on the following recommendations:
- Zero Blunt‱ TOPO‱ Donor clones (this step), 2 - 4 colonies per unique ligation
- Golden Gate clones (later steps), 4 - 6 colonies per unique ligation
5m
Preparing reagents for colony PCR.
Bring stock oligos M13F and M13R to 100 µM. In fresh, labeled 1.5 mL tubes, make 10 µM working solutions (1:10 dilution) of primers. Working solutions can be stored at -20 °C for future use.
5m
Prepare the colony PCR mixture according to the table below. It is highly recommended to make a batch reaction mix (Master Mix) for efficient, error-free set-up of multiple colony PCR reactions. Multiply the volume of each reagent by the number of colonies (n) to be screened plus one to account for pipetting error (n + 1).
| A | B | C | D | |
| Reagent | Volume per colony (µL) | Master Mix | Final Concentration | |
| M13F (10 µM) | 1.0 | 1.0 x (n+1) | 0.5 µM | |
| M13R (10 µM) | 1.0 | 1.0 x (n+1) | 0.5 µM | |
| DreamTaq Green PCR Master Mix (2X) | 10.0 | 10.0 x (n+1) | 1X | |
| MB H2O | 8.0 | 8.0 x (n+1) | ||
| TOTAL | 20.0 |
10m
Colony PCR procedure.
Label the bottom surface of an LB agar plate (with the appropriate selection antibiotic) with a numbered grid (labeled 1, 2, 3 … n) to accommodate streaks for all colonies that will be screened. Incubate the plate at 37°C until you are ready to use it in Step 7.3.
5m
Pipette 20 µL of PCR Master Mix into one PCR tube per colony (labeled 1, 2, 3 … n).
2m
Pick a colony from the ligation plate with a clean sterile micropipette tip or inoculation needle, streak the colony on the agar plate in a numbered square within the grid, and gently swirl the same tip or inoculation needle in a corresponding numbered PCR tube with Master Mix. Repeat this step with a clean tip or needle for all other colonies. Incubate the streak plate at 37°C overnight to allow the bacteria to grow.
15m
Place the PCR reactions in a thermocycler and run the following program.
| A | B | C | D | E | |
| Cycle Number | Denature | Anneal | Extend | Hold | |
| 1 | 95°C, 5 min | ||||
| 2-36 | 95°C, 15 s | 52°C, 15 s | 72°C, 30 s | ||
| 37 | 72°C, 3 min | ||||
| 38 | 4°C, infinity |
50m
Agarose gel electrophoresis. Cast a 1% (wt/vol) low EEO agarose gel in 1X TAE buffer with 1:10,000 SYBR Safe DNA Gel Stain. Load 3 µL of GeneRuler 1 kb DNA ladder in the first well and 5 µL of each colony PCR product in the remaining wells. Run the gel at 110 V for 30 - 45 minutes. If gel resolution needs to be improved, another gel can be run using increased or decreased sample loading volumes if bands are too faint or too bright.
50m
Gel Imaging and confirmation of colony PCR amplicons. Remove the gel from the electrophoresis chamber and use an imager to record the results. Compare the observed results to the expected length(s) of the PCR products. Positive clones should yield a single product the length of the donor fragment plus the distances of the primers to the cloning site. Bacteria from agar streaks for confirmed clones will be used to inoculate new liquid cultures the next day.
5m
Recombinant Plasmid Purification and Verification with Restriction Digests
9h
Liquid cultures of transformed bacterial colonies.
If colony PCR was performed, obtain the streak plate and identify the streaks that correspond with successful colony PCR results. Prepare 14 mL liquid culture tubes for the colonies to be grown by pipetting 3 mL of LB-Ampicillin (100 µg/mL) or LB-kanamycin (50 µg/mL) growth medium into each tube depending on the antibiotic resistance gene in the plasmid. Pick bacteria from the streaks with sterile pipette tips and place the tips into their respective tubes. Let the tips remain in the media during incubation; there is no need to remove them. Place tubes on a shaking rack in a 37°C incubator for at least 7 hours.
7h
For all other cloning, obtain the colony plate, use a sterile pipette tip to pick and streak one colony into a labeled, pre-warmed agar plate, and use the same tip to inoculate 3 mL of growth medium with the appropriate antibiotic. Repeat this step for 1 to 3 more colonies. The streak plate will serve as a “back-up” for subsequent liquid cultures, if needed.
Plasmid DNA extraction. Isolate the plasmid DNA from the cultures by using a plasmid miniprep kit (see Materials for the recommended kit) according to the manufacturer’s instructions.
30m
Restriction digest of plasmid DNA minipreps. For immediate verification of successful ligations, digest a small amount of each miniprep according to Table 2 and the following reaction table. For efficient, error-free set-up of several reactions, prepare a restriction digest Master Mix. Multiply the volume of each reagent by the number of minipreps to be screened plus one (n + 1). Pipette 13 µL of the restriction digest Master Mix into 0.5 mL PCR tubes. Tap the tubes gently to mix and incubate the reactions at 37°C for 5 - 30 minutes.
| A | B | C | |
| DNA Plasmid | FastDigest Enzyme(s) | Empty vector fragment(s) | |
| Donor plasmid (Zero Blunt™ TOPO™) | EcoRI | 3501 bp (backbone), 18 bp | |
| K-mer array in GGDest5-Amp | EcoRI, BcuI (SpeI) | 3245 bp (backbone), 57 bp | |
| K-mer array +BbsI in GGDest5-Amp | EcoRI, BpiI (BbsI) | One band |
TABLE 2 | FastDigest restriction enzymes for verifying recombinant plasmids. For Donor plasmids and 4x, 8x, or 12x k-mer GGDest5-Amp plasmids, successful ligations should show a band that is the length of the backbone, and a band that is the length of the desired insert plus the shorter fragment. For 4x, 8x, or 12x k-mer +BbsI GGDest5-Amp, successful 2x BbsI drop-ins should show two visible bands, whereas failed ligations that lack the 2x BbsI site will only be cut once with EcoRI.
| A | B | C | D | |
| Reagent | Volume per Miniprep (µL) | Master Mix | Final Concentration | |
| Miniprep DNA | 2.0 | 0 | (50 - 500 ng) | |
| FastDigest Green Buffer (10X) | 1.5 | 1.5 x (n+1) | 1X | |
| *Enzyme(s) | 2.0 | 2.0 x (n+1) | ||
| MB H2O | 9.5 | 9.5 x (n+1) | ||
| Total | 15.0 | 13.0 x (n+1) |
*If using one enzyme instead of two, use 1.0 µL enzyme and 1.0 μL MB H2O.
40m
Gel electrophoresis of digested DNA. After the restriction digest is complete, run the digest on a gel to verify the length of the sequence ligated into the vector. Cast a 1% (wt/vol) agarose gel in TAE buffer with SYBR DNA gel stain. Load 3 µL of GeneRuler 1 kb DNA ladder into the first well and 15 µL of digest product into the next wells. Run the gel at 100 V for 45 minutes. Shorter run times are recommended for digests that are expected to generate short (< 300 bp) bands.
50m
Validation of plasmids via Sanger Sequencing. Verify the sequence of the isolated plasmids using the primer appropriate for the vector. See Materials for primer sequences:
Donor plasmid (Zero Blunt‱ TOPO‱): Forward read, M13R; reverse read, M13F
4x, 8x, or 12x k-mer GGDest5-Amp: Forward read, M13F; reverse read, M13R
4x, 8x, or 12x k-mer +BbsI GGDest5-Amp: Forward read, M13F; reverse read, M13R
The resulting sequences should be compared to a reference sequence for the expected plasmid product.
Assembly of a 4x K-mer Array in the GGDest5-Amp Destination Vector
23h 5m
Preparation of donors and destination vector for single-pot assembly. The Golden Gate cloning method for single-pot assembly requires a 1:1 molar ratio of each insert donor to the destination vector, with a recommended amount of 1 μL each for the reaction.
Use a spectrophotometer to measure the concentrations (ng/µL) of donor and destination vector DNA stock.
10m
Determine the starting molarity in fmol/µL for each stock DNA:
Calculated DNA fmol/µL = DNA ng/µl * 10^6 / ((length of DNA bp * 617.96 ng/nmol/bp) + 36.04 ng/nmol))
Note: This formula is based on the New England Biolabs NEBioCalculator tool for converting double stranded DNA length to mass (https://nebiocalculator.neb.com/#!/dsdnaamt).
Make a working solution of 10 - 50 µL at 40 fmol/µL for each donor DNA and destination vector as shown for the examples in the table below. Avoid using stock DNA where the molarity is below 40 fmol/µL. In these cases (e.g. the Destination Vector in the example below), you may use a non-diluted volume that contains 40 fmol. Keep in mind that the total volume for all DNA components in the Golden Gate reaction is limited to 9.0 µL.
| A | B | C | D | E | F | |
| Sample | Stock DNA | Working solution (20 µL) | ||||
| Length (bp) | ng/µL | Calculated DNA fmol/µL | DNA µL | MB H2O µL | ||
| Donor Module A | 3593 | 169.0 | 76.1 | 10.5 | 9.5 | |
| Donor Module B | 3593 | 155.0 | 69.8 | 11.5 | 8.5 | |
| Donor Module C | 3593 | 153.8 | 69.3 | 11.5 | 8.5 | |
| Donor Module D | 3593 | 125.5 | 56.5 | 14.2 | 5.8 | |
| Destination Vector | 3506 | 49.0 | 22.6 | (do not dilute, use 1.8) | N/A |
5m
Golden Gate reaction set-up. Prepare the reaction(s) according to the table below, with a final volume of 10 µL per reaction.
| A | B | C | |
| Reagent | Volume (μL) | Final Concentration or Amount | |
| GGDest5-Amp | 1.0 | 40 fmol | |
| DNA Module A | 1.0 | 40 fmol | |
| DNA Module B | 1.0 | 40 fmol | |
| DNA Module C | 1.0 | 40 fmol | |
| DNA Module D | 1.0 | 40 fmol | |
| T4 DNA Ligase Buffer with 10 mM ATP (10X) | 1.0 | 1X Buffer, 1 mM ATP | |
| T4 Ligase (NEB Quick Ligase) | 0.5 | ||
| FastDigest BpiI (BbsI) | 0.5 | ||
| MB H2O | 3.0 | ||
| Total | 10.0 |
5m
Golden Gate assembly reaction. Run the single-pot assembly reaction with the following thermocycler program:
| A | B | C | D | |
| Cycle Number | Digestion | Ligation | Heat Inactivation | |
| 1-25 | 45°C, 2 min | 16°C, 5 min | ||
| 26 | 60°C, 10 min | |||
| 27 | 80°C, 20 min |
4h
Quick transformation. Transform the Golden Gate reaction product into NEB DH5ɑ-Turbo E. coli.
Combine 5 μL of the product with 50 μL of the chemically competent E. coli cells. Incubate on ice for 10 minutes.
15m
Pipette transformed cells onto a warmed agar plate (100 μg/mL ampicillin). Spread the cells with plating beads and place in a 37°C incubator for at least 7 hours.
7h
Plasmid screening with colony PCR. Follow the steps described under “Recombinant Plasmid Screening With Colony PCR” to identify 4 - 6 candidate clones with full-length inserts.
2h 30m
Recombinant plasmid preparation and verification. Follow the steps described under “Recombinant Plasmid Preparation and Verification with Restriction Digests.” It is important to include Sanger Sequencing (one forward reaction and one reverse reaction per construct) to determine if modules A - D were assembled in the correct order. The resulting k-mer array is ready to be transferred into a mammalian expression vector. If additional k-mers are desired, complete the next step.
9h
Restoration of the BbsI Golden Gate Cloning Site After the 4x K-mer
19h 22m
Produce 2x BbsI cloning site dsOligos. You will anneal the single stranded oligos (ssOligos) “Top 2xBbsI Oligo” and “Bottom 2xBbsI Oligo” to generate a short DNA fragment that contains left and right BbsI sites, and has BcuI (SpeI) and PstI-compatible sticky overhangs.
Bring the ssOligos to 100 µM with MB H2O. Dissolve with gentle heating (50°C) and occasional vortexing for 10 minutes.
15m
Set up reactions for phosphorylation and annealing of ssOligos as shown in the table below.
| A | B | C | |
| Reagent | Volume (μL) | Final Concentration | |
| 100 μM Top 2xBbsI Oligo | 1.0 | 10 μM | |
| 100 μM Bottom 2xBbsI Oligo | 1.0 | 10 μM | |
| T4 Ligation Buffer (10X) | 1.0 | 1X | |
| T4 PNK (NEB) | 0.5 | ||
| MB H2O | 6.5 | ||
| TOTAL | 10.0 | 250X |
5m
Run the ssOligo phosphorylation and annealing reaction(s) with the thermocycler program shown in the table below.
| A | B | C | D | E | |
| Cycle Number | Phosphorylate | Deactivate PNK & Denature DNA | Anneal | Hold | |
| 1 | 37°C, 30 min | ||||
| 2 | 95°C, 5 min | ||||
| 3 | Ramp down at 5°C per min to 25°C | ||||
| 4 | 4°C, ∞ |
1h
Dilute 1 µL of the resulting 250X dsOligo solution in 249 µL MB H2O to make a 1X working solution.
2m
Linearization and dephosphorylation of the 4x K-mer GGDest5-Amp plasmid.
Prepare a restriction digest as shown in the table below. Mix and incubate the reaction at 37°C for 10 minutes to digest the DNA, and then at 75°C for 5 minutes to denature/deactivate the enzymes.
| A | B | C | |
| Reagent | Volume (μL) | Final Concentration or Amount | |
| 4x k-mer GGDest5-Amp plasmid | (up to 24.0) | 1.0 μg | |
| FastDigest Buffer (10X) | 3.0 | 1X | |
| FastDigest BcuI (SpeI) | 1.0 | ||
| FastDigest PstI | 1.0 | ||
| NEB Quick CIP | 1.0 | ||
| MB H2O | Bring final volume to 30 µL | ||
| TOTAL | 30.0 |
20m
Purify the linearized DNA using a Zymo Clean and Concentrator-25 kit, or a similar approach. An elution volume of 25 μL is recommended to yield the highest concentration.
15m
Ligation. In sterile, labeled 0.5 mL tubes, prepare a ligation and a negative control (no insert) reaction as shown in the table below, using the purified linearized 4x k-mer GGDest5-Amp plasmid (from Step 23) and the 1X 2x BbsI dsOligo (from Step 22). Incubate at room temperature for at least 10 minutes. Meanwhile, thaw chemically competent cells on ice (see the next step).
| A | B | C | D | |
| Reagent | Volume (μL) | Final Concentration or Amount | ||
| Ligation | Negative control | |||
| Linearized 4x k-mer GGDest5-Amp plasmid | (up to 7.5) | (up to 7.5) | 50 ng | |
| 2x BbsI dsOligo (1X) | 1.0 | 0 | 0.1X | |
| T4 ligase buffer (10X) | 1.0 | 1.0 | 1X | |
| NEB Quick Ligase | 0.5 | 0.5 | ||
| MB H2O | Bring final volume to 10 µL | |||
| TOTAL | 10.0 |
15m
Quick transformation. Transform the Golden Gate reaction product into a competent E. coli strain using guidelines appropriate for the strain. We recommend NEB DH5ɑ-Turbo. Quick transformation (skipping outgrowth in SOC) is possible for plasmids that carry an ampicillin resistance marker, i.e. the GGDest5-Amp backbone.
Thaw NEB DH5ɑ-Turbo E. coli cells (50 μL per reaction) on ice. Add 50 μL of thawed cells to the Ligation reaction, pipette up and down three times, and place on ice. Repeat this step for the negative control. Incubate these samples on ice for 5 minutes.
10m
Pipette the transformed cells (60 μL total) onto a warmed agar plate (100 μg/mL ampicillin). Spread the cells with plating beads and place in a 37°C incubator for at least 8 hours for DH5ɑ-Turbo cells. Other strains may require a longer incubation (overnight). A successful ligation and transformation should show < 10 colonies on the negative control plate and 5 - 10 times as many on the ligation plate.
8h
Recombinant plasmid verification. Follow the steps described under “Recombinant Plasmid Preparation and Verification with Restriction Digests” (Steps 11 - 15). It is important to include Sanger Sequencing to verify that the BbsI cloning site has no mutations.
9h
Assembly of an 8x K-mer Using the 4x K-mer +BbsI GGDest5-Amp Construct
1d 18h 35m
Complete the steps under the section “Assembly of a 4x K-mer Array in the GGDest5-Amp Destination Vector” with the following modifications.
Preparation of donors and destination vector for single-pot assembly. For the destination vector, use the 4x k-mer +BbsI GGDest5-Amp plasmid that was generated in the previous procedure “Restoration of the BbsI Golden Gate Cloning Site After the 4x K-mer”.
15m
Golden Gate reaction set-up. Follow the procedure as described.
5m
Golden Gate assembly reaction. Follow the procedure as described.
4h
Quick transformation. Follow the procedure as described.
7h 15m
Plasmid screening with colony PCR. Follow the procedure as described.
2h 30m
Recombinant plasmid verification. Follow the steps described under “Recombinant Plasmid Preparation and Verification with Restriction Digests” (Steps 11 - 15). It is important to include Sanger Sequencing (one forward reaction and one reverse reaction per construct) to determine if modules A - D were assembled in the correct order. The resulting k-mer array is ready to be transferred into a mammalian expression vector. If additional k-mers are desired, complete the next step.
9h
Restore the BbsI Golden Gate cloning site after the 8x k-mer. To prepare the plasmid for the addition of 4 more k-mers, follow the steps under “Restoration of the BbsI Golden Gate Cloning Site After the 4x K-mer” to generate a 8x K-mer +BbsI GGDest5-Amp construct.
19h 30m
Assembly of a 12x K-mer Using the 8x K-mer +BbsI GGDest5-Amp Construct
1d 18h 35m
Complete the steps under the section “Assembly of a 4x K-mer Construct in the GGDest5-Amp Destination Vector” with the following modifications.
Preparation of donors and destination vector for single-pot assembly. Use the 8x kmer +BbsI GGDest5-Amp plasmid generated in the previous procedure “Assembly of an 8x K-mer Using the 4x k-mer GGDest5-Amp Construct” as the destination vector.
15m
Golden Gate reaction set-up. Follow the procedure as described.
5m
Golden Gate assembly reaction. Follow the procedure as described.
4h
Quick transformation. Follow the procedure as described.
7h 15m
Plasmid screening with colony PCR. Follow the procedure as described.
2h 30m
Recombinant plasmid verification. Follow the steps described under “Recombinant Plasmid Preparation and Verification with Restriction Digests” (Steps 11 - 15). It is important to include Sanger Sequencing (one forward reaction and one reverse reaction per construct) to determine if modules A - D were assembled in the correct order. The resulting k-mer array is ready to be transferred into a mammalian expression vector. If additional k-mers are desired, complete the next step.
9h
Optional: Restore the BbsI Golden Gate cloning site after the 12x k-mer. To prepare the plasmid for the addition of 4 more k-mers, follow the steps under “Restoration of the BbsI Golden Gate Cloning Site After the 4x K-mer” to generate a 12x K-mer +BbsI GGDest5-Amp construct.
19h 30m
Transfer of K-mer Arrays from GGDest5-Amp into a Mammalian Expression Vector
22h 50m
Digestion of the mammalian expression vector backbone. Select one of the expression vectors from Table 3 based on the discussion in the Guidelines. Prepare a digestion reaction based on the reaction table. Incubate at 37 °C for 5 to 30 minutes. If the reaction includes phosphatase, deactivate the enzyme by incubating the reaction at 80 °C for 2 minutes.
| A | B | C | D | |
| Vector | Size (bp) | FastDigest Enzyme(s) | Phosphatase | |
| pcDNA FRT TOPO | 5137 | NotI | NEB Quick CIP | |
| pSBtetTA-YP_NotISpeI | 6407 | NotI, BcuI (SpeI) |
TABLE 3 | Mammalian expression vectors for the expression of synthetic lncRNAs.
| A | B | C | |
| Reagent | Volume (μL) | Final Concentration or Amount | |
| DNA | 0.5 - 1.0 μg | ||
| *Enzymes(s) | 2.0 | ||
| 10X FastDigest buffer | 3.0 | 1X | |
| MB H2O | Bring final volume to 30 µL | ||
| TOTAL | 30.0 |
*For pcDNA FRT TOPO use 1.0 μL NotI and 1.0 μL Quick CIP. For pSBtetTA-YP_NotISpeI use 1.0 μL NotI and 1.0 μL BcuI (SpeI).
40m
Digestion of the k-mer array insert. Digest the 4x, 8x, or 12x k-mer GGDest5-Amp plasmid with NotI and SpeI according to the table below. Incubate at 37 °C for 5 to 30 minutes.
| A | B | C | |
| Reagent | Volume (μL) | Final Concentration or Amount | |
| DNA | 0.5-0.75 µg | ||
| FastDigest NotI | 0.5 | ||
| FastDigest BcuI (SpeI) | 0.5 | ||
| 10X FastDigest buffer | 3.0 | 1X | |
| MB H2O | Bring final volume to 30 µL | ||
| TOTAL | 30.0 |
40m
Gel purification of the backbone and insert fragments.
Cast a 0.8% (0.48 g/ 60 mL) low EEO agarose gel in 1X TAE buffer with 1:10,000 SYBR Safe DNA Gel Stain. Use a comb with extra large wells that can accommodate 30 µL. Load 6 µL of GeneRuler 1 kb DNA ladder in the first well and 30 µL of each digested insert fragments or expression vector fragments in the remaining wells. Run the gel at 110 V for 30 - 45 minutes.
50m
Place the gel with the digested expression vector on a blue-light transilluminator. We use the E-Gel‱ Power Snap Electrophoresis System (Invitrogen). If the gel is too big to fit into a transilluminator, trim the gel with scalpel while not disturbing the bands that contain DNA fragments.
5m
Wear an eye protector that filters the blue light from the transilluminator. We use Safe Imager‱ Viewing Glasses (Invitrogen). Then turn on the blue-light on the transilluminator. The DNA bands should now be visible. Use the scalpel to isolate the target DNA fragment that contains the linearized backbone of the expression vector. Use eye protection to minimize your exposure to the blue light.
10m
Transfer the gel slice into labelled 1.5 mL microcentrifuge tubes. Extract the DNA from the gel slices using a DNA gel extraction kit (e.g. NEB Monarch DNA Gel Extraction Kit Protocol) following the manufacturer’s instructions. Elute with 20 µL of pre-warmed elution buffer.
30m
Measure the concentration of each purified product with a spectrophotometer.
5m
Ligation of DNA Fragments. Set up ligation reactions with the k-mer array insert and without the insert (negative control) based on the table below. Mix 20 - 50 ng of vector backbone with 3:1 molar ratio of insert (k-mer array) to vector. Use the following formula to calculate the required volume of insert:
Insert fragment volume = (Amount of Vector Backbone (ng) × Insert Size (bp)) / (Vector Size (bp) × Insert:Vector Molar Ratio × Insert Concentration (ng/μL))
Incubate the reaction at room temperature for 10 minutes.
| A | B | C | D | |
| Reagent | Volume (μL) | Final Concentration or Amount | ||
| Ligation | Negative control | |||
| K-mer array insert | 0 | Insert 1:3 Backbone vector molar ratio | ||
| Linearized Vector Backbone | 20 - 50 ng | |||
| 10X T4 DNA Ligase Reaction Buffer (NEB) | 1.0 | 1.0 | 1X | |
| T4 DNA Ligase (NEB) | 1.0 | 1.0 | ||
| MB H2O | Bring final volume to 10 µL | |||
| TOTAL | 10.0 | 10.0 |
15m
Transformation of ligated products: Add 10 µL of the ligation reaction to 50 µL of competent DH5α-Turbo cells. Incubate the cells on ice for 5 minutes. Plate 50 µL of the transformation mixture onto pre-warmed LB agar plates containing ampicillin and spread using beads. Incubate the mixtures overnight at 37°C.
8h
Plate inspection. Obtain colony plates from the incubator and inspect for colony growth.
Expect to see multiple colonies on the ligation plates and significantly fewer colonies on the negative control plate
5m
Colony PCR. Complete the steps under the section “Recombinant Plasmid Screening with Colony PCR” (Steps 5 - 10).
2h 30m
Recombinant Plasmid Purification and Verification with Restriction Digests. Complete the steps under the section “Recombinant Plasmid Purification and Verification with Restriction Digests” (Steps 11 - 15) with the following modifications.
Liquid cultures of transformed bacterial colonies. Follow the procedure as described.
7h
Plasmid DNA extraction. Follow the procedure as described.
30m
Restriction digest of plasmid DNA minipreps. Follow the procedure as described but with the modified Table 4 below
| A | B | C | |
| Vector | FastDigest Enzyme(s) | Empty vector fragment(s) (bp) | |
| K-mer array in pcDNA FRT TOPO | EcoRI | 4258, 879 | |
| K-mer array in pSBtetTA-YP_NotISpeI | EcoRI, BcuI (SpeI) | 4843, 1564 |
TABLE 4 | FastDigest restriction enzymes for verifying recombinant plasmids. For pcDNA FRT TOPO-K-mer plasmid, successful transfer of the K-mer fragment should show a band with the length 4258 bp, and a band with the length 879 bp plus the length of the K-mer insert. For pSBtetTA-YP_NotISpeI-K-mer, successful transfer of the K-mer fragment should show a band with the length 4843 bp plus the length of the K-mer insert, and a band with the length 1564 bp. This digestion assumes that the K-mer sequence does not contain any of the EcoRI or the BcuI (SpeI) sites.
| A | B | C | |
| Reagent | Volume per Miniprep (µL) | Final Concentration or Amount | |
| Miniprep DNA | 0.5 µg | ||
| FastDigest Green Buffer (10X) | 1.5 | ||
| *Enzyme(s) | 2.0 | ||
| MB H2O | Bring final volume to 15 µL | ||
| Total | 15.0 |
* For pcDNA FRT TOPO-K-mer use 1.0 μL EcoRI and 1.0 μL MBH2O. For pSBtetTA-YP_NotISpeI-K-mer use 1.0 μL EcoRI and 1.0 μL BcuI (SpeI).
40m
Gel electrophoresis of digested DNA. Follow the procedure as described.
50m
Validation of plasmids via Sanger Sequencing. Verify the sequence of the isolated plasmids by sequencing using a primer that is appropriate for the vector. See Materials for primer sequences:
- pcDNA FRT TOPO-K-mer: Forward read, CMV-Forward; reverse read, BGHR
- pSBtetTA-YP_NotISpeI-K-mer: Forward read, pSBtetTA-YP_F; reverse read, pSBtetTA-YP_R
The resulting sequences should be compared to a reference sequence for the expected plasmid product.
Protocol references
1. Mattick JS, Amaral PP, Carninci P, Carpenter S, Chang HY, Chen L-L, et al. Long non-coding RNAs: definitions, functions, challenges and recommendations. Nature Reviews Molecular Cell Biology. 2023;24: 430–447.
2. Mercer TR, Mattick JS. Structure and function of long noncoding RNAs in epigenetic regulation. Nature Structural & Molecular Biology. 2013;20: 300–307.
3. Cabili MN, Dunagin MC, McClanahan PD, Biaesch A, Padovan-Merhar O, Regev A, et al. Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution. Genome Biology. 2015;16: 1–16.
4. Sunwoo H, Wu JY, Lee JT. The Xist RNA-PRC2 complex at 20-nm resolution reveals a low Xist stoichiometry and suggests a hit-and-run mechanism in mouse cells. Proceedings of the National Academy of Sciences. 2015;112: E4216–E4225.
5. Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434: 400–404.
6. Ross MT, Grafham DV, Coffey AJ, Scherer S, McLay K, Muzny D, et al. The DNA sequence of the human X chromosome. Nature. 2005;434: 325–337.
7. Unfried JP, Ulitsky I. Substoichiometric action of long noncoding RNAs. Nature Cell Biology. 2022;24: 608–615.
8. Gao F, Cai Y, Kapranov P, Xu D. Reverse-genetics studies of lncRNAs—what we have learnt and paths forward. Genome Biology. 2020;21: 1–23.
9. Dong A, Preusch CB, So W-K, Lin K, Luan S, Yi R, et al. A long noncoding RNA, LncMyoD, modulates chromatin accessibility to regulate muscle stem cell myogenic lineage progression. Proceedings of the National Academy of Sciences. 2020;117: 32464–32475.
10. Grossi E, Raimondi I, Goñi E, González J, Marchese FP, Chapaprieta V, et al. A lncRNA-SWI/SNF complex crosstalk controls transcriptional activation at specific promoter regions. Nature Communications. 2020;11: 1–16.
11. Pistoni M, Rossi T, Donati B, Torricelli F, Polano M, Ciarrocchi A. Long Noncoding RNA Acts as a Molecular Switch for BRD4 Transcriptional Activity and Mediates Repression of BRD4/WDR5 Target Genes. Mol Cancer Res. 2021;19: 799–811.
12. Much C, Lasda EL, Pereira IT, Vallery TK, Ramirez D, Lewandowski JP, et al. The temporal dynamics of lncRNA Firre-mediated epigenetic and transcriptional regulation. Nature Communications. 2024;15: 1–13.
13. Bonilla SL, Jones AN, Incarnato D. Structural and biophysical dissection of RNA conformational ensembles. Curr Opin Struct Biol. 2024;88: 102908.
14. Duszczyk MM, Wutz A, Rybin V, Sattler M. The Xist RNA A-repeat comprises a novel AUCG tetraloop fold and a platform for multimerization. RNA. 2011;17: 1973–1982.
15. Ilik IA, Quinn JJ, Georgiev P, Tavares-Cadete F, Maticzka D, Toscano S, et al. Tandem stem-loops in roX RNAs act together to mediate X chromosome dosage compensation in Drosophila. Mol Cell. 2013;51: 156–173.
16. Sentürk Cetin N, Kuo C-C, Ribarska T, Li R, Costa IG, Grummt I. Isolation and genome-wide characterization of cellular DNA:RNA triplex structures. Nucleic Acids Res. 2019;47: 2306–2321.
17. Kuo C-C, Hänzelmann S, Sentürk Cetin N, Frank S, Zajzon B, Derks J-P, et al. Detection of RNA-DNA binding sites in long noncoding RNAs. Nucleic Acids Res. 2019;47: e32.
18. Zhao Z, Sentürk N, Song C, Grummt I. lncRNA PAPAS tethered to the rDNA enhancer recruits hypophosphorylated CHD4/NuRD to repress rRNA synthesis at elevated temperatures. Genes Dev. 2018;32: 836–848.
19. Brown JA, Bulkley D, Wang J, Valenstein ML, Yario TA, Steitz TA, et al. Structural insights into the stabilization of MALAT1 noncoding RNA by a bipartite triple helix. Nat Struct Mol Biol. 2014;21: 633–640.
20. Kirk JM, Kim SO, Inoue K, Smola MJ, Lee DM, Schertzer MD, et al. Functional classification of long non-coding RNAs by k-mer content. Nat Genet. 2018;50: 1474–1482.
21. Fan Y, Chen M, Zhu Q. lncLocPred: Predicting LncRNA Subcellular Localization Using Multiple Sequence Feature Information. [cited 18 Dec 2024]. Available: https://doi.org/10.1109/ACCESS.2020.3007317
22. Shi K, Liu T, Fu H, Li W, Zheng X. Genome-wide analysis of lncRNA stability in human. PLoS Comput Biol. 2021;17: e1008918.
23. Colognori D, Sunwoo H, Kriz AJ, Wang C-Y, Lee JT. Xist Deletional Analysis Reveals an Interdependency between Xist RNA and Polycomb Complexes for Spreading along the Inactive X. Mol Cell. 2019;74: 101–117.e10.
24. Minks J, Baldry SEL, Yang C, Cotton AM, Brown CJ. XIST-induced silencing of flanking genes is achieved by additive action of repeat a monomers in human somatic cells. Epigenetics & Chromatin. 2013;6: 1–10.
25. Nakamoto MY, Lammer NC, Batey RT, Wuttke DS. hnRNPK recognition of the B motif of Xist and other biological RNAs. Nucleic Acids Res. 2020;48: 9320–9335.
26. Kratz MB, Smith KN. Predicting conserved functional interactions for long noncoding RNAs via deep learning. Front RNA Res. 2024;2: 1473293.
27. Wutz A, Rasmussen TP, Jaenisch R. Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nature Genetics. 2002;30: 167–174.
28. Navarro-Cobos MJ, Morales-Guzman SI, Baldry SEL, Brown CJ. Derivation of a minimal functional XIST by combining human and mouse interaction domains. Hum Mol Genet. 2023;32: 1289–1300.
29. Yao Y, Jin S, Long H, Yu Y, Zhang Z, Cheng G, et al. RNAe: an effective method for targeted protein translation enhancement by artificial non-coding RNA with SINEB2 repeat. Nucleic Acids Res. 2015;43: e58.
30. Cao C, Li A, Xu C, Wu B, Liu J, Liu Y. Enhancement of protein translation by CRISPR/dCasRx coupled with SINEB2 repeat of noncoding RNAs. Nucleic Acids Res. 2023;51: e33.
31. Shechner DM, Hacisuleyman E, Younger ST, Rinn JL. Multiplexable, locus-specific targeting of long RNAs with CRISPR-Display. Nat Methods. 2015;12: 664–670.
32. Brockdorff N. X-chromosome inactivation: closing in on proteins that bind Xist RNA. Trends Genet. 2002;18: 352–358.
33. Fernandez-Moreno J-P, Yaschenko AE, Neubauer M, Marchi AJ, Zhao C, Ascencio-Ibanez JT, et al. A rapid and scalable approach to build synthetic repetitive hormone-responsive promoters. Plant Biotechnol J. 2024;22: 1942–1956.
34. Haynes KA, Priode JH. Rapid Single-Pot Assembly of Modular Chromatin Proteins for Epigenetic Engineering. Methods Mol Biol. 2023;2599: 191–214.
35. GGDest5-Amp · Benchling. [cited 24 Jan 2025]. Available: https://benchling.com/s/seq-kwIQ58J5GurG5CJWGzPJ?m=slm-GvgfysuZdMMUtLBYlIb4
36. Kowarz E, Löscher D, Marschalek R. Optimized Sleeping Beauty transposons rapidly generate stable transgenic cell lines. Biotechnol J. 2015;10: 647–653.
37. pSBtetTA-YP_NotISpeI · Benchling. [cited 24 Jan 2025]. Available: https://benchling.com/s/seq-TonJUz4q5bOm0RAfWLGl?m=slm-nbYdBYVn0Mp5rueQhAbq
38. Ivics Z, Hackett PB, Plasterk RH, Izsvák Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91: 501–510.
Acknowledgements
The development of this protocol was supported by award NSF NSF 2243665 to K. Haynes.