Mar 29, 2023
Benchmarking protocol for plant genomes
- Vidya Vuruputoor1,
- Daniel Monyak1,
- Karl C Fetter1,
- Akriti Bhattarai1,
- Bikash Shrestha1,
- Sumaira Zaman1,
- Jeremy Benett1,
- Susan L McEvoy1,
- Madison Caballero1,
- Jill L Wegrzyn1,
- Cynthia Webster1
- 1UConn
- PlantCompGenomics



Protocol Citation: Vidya Vuruputoor, Daniel Monyak, Karl C Fetter, Akriti Bhattarai, Bikash Shrestha, Sumaira Zaman, Jeremy Benett, Susan L McEvoy, Madison Caballero, Jill L Wegrzyn, Cynthia Webster 2023. Benchmarking protocol for plant genomes. protocols.io https://dx.doi.org/10.17504/protocols.io.yxmvmkp95g3p/v1
License: This is an open access protocol distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Protocol status: Working
We use this protocol and it's working
Created: February 19, 2021
Last Modified: March 29, 2023
Protocol Integer ID: 47436
Keywords: several plant genome, de novo annotation software like braker, benchmarking protocol, using de novo annotation software, benchmarking stats visit, seq data of the target species, protocol for plant, genome sequence, genome, read rna, rna, more stats on the runs visit, target species, particular species, plant
Funders Acknowledgements:
Diversity of Physcomitrium pyriforme in North America and Europe: significance of autopolyploidy within a phylogenomic and experimental framework
Grant ID: DEB-1753811
EASEL
Grant ID: DBI-1943371
Abstract
This is the protocol we used to benchmark several plant genomes. We first gather all available information of a particular species, from genome sequence to short-read and long-read RNA-seq data of the target species. Then, we annotate the genome by using de novo annotation software like BRAKER and MAKER.
Manuscript link: https://www.biorxiv.org/content/10.1101/2022.10.03.510643v3
For scripts used:
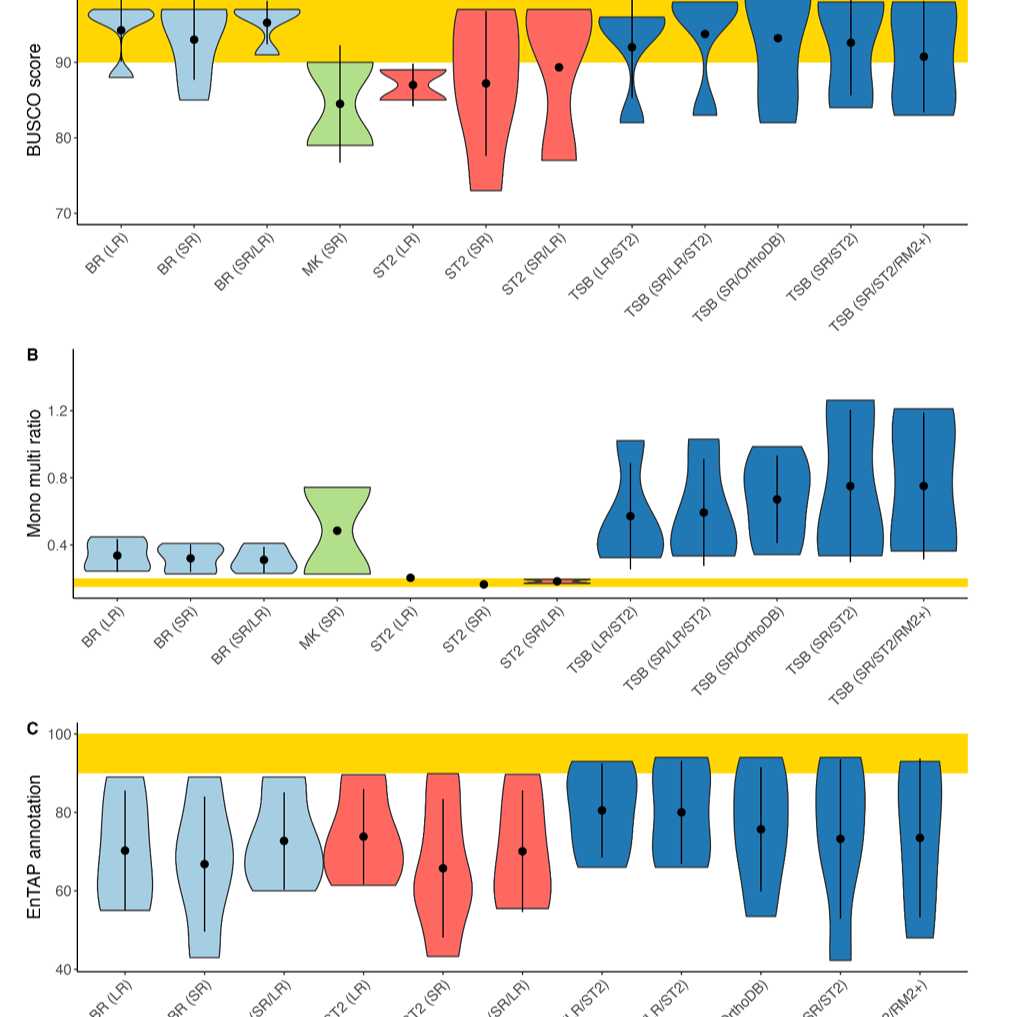
For benchmarking stats visit:
For more stats on the runs visit:
Guidelines
Materials
Visit https://gitlab.com/PlantGenomicsLab/annotationtool/-/tree/master/data-gathering for the scripts and a detailed summary of all the steps followed in benchmarking plant genomes
Before start
Place all files relevant to a step in the same directory. For example, all files related to genome evidence (under step 1) are placed in a directory "genome_evidence"
Download/gather genome resources
- Unmasked genome (for MAKER-P) pipeline
- Soft-masked genome (if available)
- gff, gtf files
- cDNA and pep files
Note
Please note the version of the genome resource.
Filter genome
1. filtersubmit.sh removes sequences less than 500 bp
- The script filtersubmit.sh calls filterLen.py (custom script, no version no.)
Safety information
Make sure to filter the genome of <500 bp scaffolds before masking the genome
Overnight This step could take some time depending on the size of the genome
Soft-mask genome
Note
Check if genome is soft-masked first. If not, proceed with masking step.
1. Run repeatModeler.sh which loads RepeatModeler/2.01 to make the repeat library
Safety information
Run the genome with and without LTRStruct (these are two different RepeatModeler scripts)
Command
RepeatModeler2 generates repeat libraries which would be masked in the subsequent RepeatMasker step. The option, -LTRStruct helps to find more LTR components in the genome.
RepeatModeler2
1a. (optional) Run splitfasta.sh which loads anaconda and breaks the filtered genome into 100 pieces
Safety information
RepeatMasker can be run on the whole genome at once or on pieces of the genome separately using a SLURM array.
If splitting up the genome, run the splitfasta.sh script first.
2. Run repeatmasker.sh which loads RepeatMasker/4.0.6 Important: Check version before running this program.
If splitting up the genome, run repeatmasker_array.sh
If running on the genome as a whole, run repeatmasker.sh
Command
RepeatMasker actually masks the repeats generated by RepeatModeler
RepeatMasker
3. Calculate repeat content
3a. If RepeatMasker was run on the genome all at once (repeatmasker.sh), look at the repeat content value in the .tbl file
EXAMPLE:
Note
==================================================
file name: Arabidopsis_thaliana.TAIR10.dna.toplevel.fa
sequences: 7
total length:119667750 bp(119482146 bp excl N/X-runs)
GC level: 36.06 %
bases masked: 18219891 bp ( 15.23 %)
3b. If RepeatMasker was run as a SLURM array (split genome), use total_pct_masked.sh script.
Calculates correct repeat content fraction using all the .tbl files.
Provide path of directory with .tbl files as command line input with script.
Outputs FRACTION of genome that is masked.
4. Verify repeat content
rep_content.sh calculates repeat content.
Uses python script rep_content.py
Provide genome file as command line input with script.
Can be used to calculate repeat content for a genome that you did not mask yourself, as well as verifying repeat content seen in a RepeatMasker .tbl file
Outputs FRACTION of genome that is masked.
Genome quality assessment
1. Check quality of the filtered genome with Quast (quast.sh calls quast 5.0.2)
Command
QUAST
2. Check completeness of the filtered genome with BUSCO (busco.sh calls busco/4.1.2)
Command
BUSCO
Safety information
Have AUGUSTUS installed in a writeable directory
Creating indices for the alignment of evidence (RNA-Seq data and transcriptome) to the genome
Hisat2 and Gmap are used for aligning RNA-Seq (step 6) and transcriptome data (step 7), respectively.
Prior to the alignments, indices of the genome need to be created.
Gmap index
- gmap_index.sh calls gmap/2019-06-10 gmap_build
- -D # sets the output directory
- -d sets the index prefix
Command
gmapBuild
- pass in the masked, filtered genome fasta file last
- output files end in .chromsome, .chromosome.iit, .chrsubset, .contig, .contig.iit, .genomebits128, .genomecomp, .maps, .ref153offsets64meta, .ref153offsets64strm, .ref153positions, .sachildexc, .sachildguide1024, .saindex64meta, .saindex64strm, .salcpchilddc, .salcpexc, .salcpguide1024, .sarray, .version
Hisat index
- hisatBuild.sh calls hisat2/2.2.0 hisat-build
- -f sets the input .fasta file
- set the output directory/prefix last
Command
Hisat Build
- output files are incrementing numbers like so: Arabidopsis_genome.1.ht2
Gather evidence
Go to NCBI to fetch short-read and long-read data from RNA-seq studies of the species
Prepare the short-read RNAseq data
1. Use the trinityfetchSRA.sh script to get reads from NCBI.
- Downloads FASTQ files with header format that can be used for hisat2 read alignment as well as Trinity transcriptome assembly
- fastq-dump --defline-seq '@$sn[_$rn]/$ri' --split-files $LINE
- Option —-split-fastq to separate pairs into their respective left and right files while fetching.
Safety information
- at least 10M reads (spots) and >90% mapping rate for each library used for support
- The library should be RNA-Seq based and not focused on plastid (chloroplast or mitochondria)
- Make a .txt file containg the SSR numbers of the reads to download.
- trinityfetchSRA.sh calls sratoolskit/2.8.1
- In trinityfetchSRA.sh, change the FILENAME to match your .txt.
- output is .fastq files in the same directory.
- move .fastq files to organism
2. Validate the retrieved reads using validateSRA.sh
- validateSRA.sh calls sratoolskit/2.8.1
- FILENAME # sets input file
- output is logged to *.err
- move reads to raw_reads dir
3. Trim reads using sickle
- sickle.sh calls sickle/1.33
- rawreadsdir # sets input dir
- output is .fastq files, two for each pair (1 and 2) that begin with trimmed plus single_trimmed for singles created by sickle.
4. Check quality of trimmed reads
- fastqc.sh loads fastqc/0.11.7
Safety information
Do take the time to check the FASTQC reports, this serves as a sanity check of whether the reads are good enough to use.
Pay attention to "Adaptor Content" and "Overrepresented Sequences" (may show adaptor sequences)
If there is signifcant adaptor content, go back to the raw read and trim this content using a tool like Scyte, Trimmomatic, or TrimGalore. Then, redo the Sickle trimming (except if using TrimGalore)
- readsdir # input directory
- output *_fastqc.html and *_fastqc.zip for each run
Safety information
Do hisat2 alignment (step 6) before creating Trinity de novo transcriptome
After hisat2 alignment, alignRates.sh (step 6.1.5) will show which libraries have an alignment (mapping) rate >= 90%
Only use the libraries with alignment rate >= 90% for Trinity de novo transcriptome
Align short-read (RNA-Seq) to the genome
- Run the hisat2 aligner
- hisat.sh calls hisat2/2.2.0
- orgdir/trimmeddir # set input directory (dir containing trimmed fastq)
- output is .sam files, one for each run (SRR file)
1.5. Run alignRates.sh
- Outputs files showing each hisat2 alignment (mapping) rate for each SRA library
- If rate >= 90%, it prints the SRA accession to good_SRAs.txt and prints the rate to good_rates.txt
Safety information
Only use libraries with alignment rate >= 90%
Only use good libraries to create BAM file with samtools.sh in next step
Only use good libraries to do Trinity transcriptome assembly
2. Convert, sort, and merge the human-readable .sam file to compressed machine-readable .bam
- samtools.sh calls samtools/1.7
- input location of .sam files
- output is one file with .bam extension
Safety information
samtools_updated.sh makes this process easier by only converting, sorting, and merging libraries in good_SRAs.txt (i.e. had a mapping rate >= 90%)
Assemble transcriptome and align to the genome
Safety information
Do NOT use a TSA from NCBI.
For consistency, all short-read-based transcriptomes will be created de novo with Trinity
- Create de novo transcriptome with short reads and Trinity
- Use trimmed short-read libraries from before
Safety information
Run trinity separately on all libraries, and then concatenate
- Concatenate runs where necessary if they are from the same library, but just different lanes. Lanes are just a byproduct of the ways things are separated during the sequencing process, but it’s all a part of the same sequencing library. Sometimes these are loaded into NCBI as separate runs. Library names can be found on the experiment (SRX) page. Lane info can be seen sometimes on the SRX page or by clicking on the SRR id links from the experiment page to go to the SRA Run Browser MetaData tab.
- Do not concatenate replicants. There are two kinds of replicants: sample and technical.
- Sample: Run these separately in Trinity.
- Technical: If a sample has multiple runs from the same library and same lane, it might be a technical replicant. If there is already enough read depth, we don’t need multiple replicants of this kind.
- If the previous details are not clear in NCBI, referring to the published study may help clarify.
Safety information
Only use libraries with alignment rate >= 90% (see hisat2 SRA alignment step)
trinity.sh calls trinity/2.8.5
- Run Trinity separately on all libraries - see trinity_updated.sh - runs as Slurm array on all libraries simultaneously
- PE_1 # sets the left read for each library
- PE_2 # sets the right read for each library
- out # sets the output directory
- min_contig_length is 300
Command
Trinity
2. add_prefixes_trinity.sh
- Adds the SRA accession to the beginning of each header in all of the Trinity FASTA assemblies
- Gives all transcripts a unique name, prevents confusion of transcripts later
- Outputs the new FASTA files in $org/evidence/transcriptome/trinity/trinity_prefix/
3. catTrinity.sh
- Concatenates all Trinity assemblies together
4. Frameselect transcriptome
- Input is file created before by catTrinity.sh - all Trinity assemblies concatenated together
- frameselect.sh runs TransDecoder/5.3.0
- clustered_TSA # sets directory that contains the TSA file
- filename # sets TSA file name
Command
Transdecoder finds the coding regions in transcript sequences.
Note: frameselect.sh requires the transcriptome to have a certain number of sequences to work (> 31)
Transdecoder
- output files are placed in directory 'bestHit' (.bed, .cds, .gff3, .pep)
5. Cluster frameselected transcriptome
- usearch.sh runs usearch/9.0.2132
- percent id is 0.98
- concat_file # sets the input file (.cds from previous step)
- output centroids_$concat_file and centroids_$concat_file.uc
6. Remove short genes (less than 300bps) from the centroids file
- Run filter300.sh which calls filterLen.py
Note
At this point, you have your final transcriptome
(the filtered centroids file)
7. Run the gmap aligner
- gmap.sh calls gmap/2019-06-10
- idx # sets the location of the index
- prefix # sets the prefix of the index files
- fasta sets the location of the frameselected, clustered, filtered TSA as input
- -a 1 # Translate codons from given nucleotide (1-based)
- --cross-species # Use a more sensitive search for canonical splicing, which helps especially for cross-species alignments and other difficult cases
- -D # sets idx
- -d # sets prefix
- -f gff3_gene # sets the format
- --fulllength # Assume full-length protein, starting with Met
- --min-trimmed-coverage=0.95 # Do not print alignments with trimmed coverage less than this value
- --min-identity=0.95 # Do not print alignments with identity less than this value
- -n1 # set maximum number of paths to show. If set to 1, GMAP will not report chimeric alignments, since those imply two paths. If you want a single alignment plus chimeric alignments, then set this to be 0.
Command
Gmap aligns proteins to the genome
gmap
- output files are $prefix_gmap.gff3 and $prefix_gmap.error
Assemble proteome and align to the genome
- Filter the .pep file created from frame selection
- run filterpep.sh to filter the proteins so that you only have the same sequences that correspond to the genes left after filtering in Step 7.3.
- filterpep.sh trims headers if there is additional info after id, makes a list of headers from filtered TSA, and then calls CreateFasta.py
- output is *_filtered.pep
Note
At this point, you have your final proteome
(the *_filtered.pep file)
2. Run the genomeThreader aligner
- genomeThreader.sh calls genomeThreader/1.7.1 and genometools/1.6.1
- gt adds stop amino acids *
- gth aligns the protein sequences
Safety information
It is common for the error file to say "warning: protein sequence "some-transcript" does not end with a stop amino acid ('*')" for one transcript.
Command
Genome Threader computes gene structure predictions.
Genome Threader
Prepare Stringtie proteins (using genome-guided aligner)
1. Run Stringtie to align reads
- 0_stringtie.sh calls Stringtie and creates a gtf file in the directory
2. Extract proteins from the alignment file
- 1_gffread.sh produces a protein file in the current directory
3. Frameselect the protein file using TransDecoder v5.5
- 2_frameselect.sh uses TransDecoder and Eggnog to frame-select proteins
Run genome annotation software
Genome annotation with BRAKER
Safety information
IMPORTANT STEPS BEFORE RUNNING BRAKER
## Copy software to home directory
cp -r /labs/Wegrzyn/annotationtool/software/BRAKER_2.1.5 ~/
cp -r /labs/Wegrzyn/annotationtool/software/Augustus_3.3.3 ~/Augustus
cp -r /labs/Wegrzyn/annotationtool/software/cdbfasta ~/
Obtaining Genemark-ET software and license key
- Select: GeneMark-ES/ET/EP ver 4.64_lic - LINUX 64
- Fill in information
- To download the software and the key, right-click on the respective link (64_bit for the key), and click "copy link address"
cd ~
# Download, unzip, and rename the key
wget "enter-key-link-here"
gzip -d gm_key_64.gz
mv gm_key_64 local_gm_key
# Genemark key may expire every few months, so go back and get a new one
# Download and unzip the software
wget "enter-software-link-here"
tar -xf gmes_linux_64.tar.gz
rm gmes_linux_64.tar.gz
# Change header in all perl scripts of the Genemark folder, fixes perl path
cd gmes_linux_64
module load perl/5.28.1
perl change_path_in_perl_scripts.pl "/usr/bin/env perl"
- Run 1: Input- short read data
- Use a new directory for this: annotaions/braker/braker.sh
- genome from species of interest
- braker.sh calls BRAKER/2.1.5, augustus, genemark, bamtools
- augustus must be in your local directory (should be there from Step 3.3)
- make a directory in your home called 'tmp'
- softmasking 1 # indicates the genome has been softmasked
- copies of perl modules from the lab directory root are included for specific dependencies
- The merged .bam file from Step 6 is used as input.
braker.pl --species=yourSpecies --genome=genome.fasta \
--bam=species.bam \
--softmasking 1 \
--gff3 \
--cores 10
Safety information
VERY IMPORTANT
Make sure to have a separate species name for each alternate BRAKER run if running at the same time.
Otherwise, species gene data in your Augustus folder can be deleted or changed without throwing an error.
2. Run 2: Input- short read data and de novo proteins from species of interest
- Use a new directory for this: annotaions/braker2/braker_prot.sh
- proteins from species of interest
- short reads from species of interest
- braker_protein.sh
- like braker.sh but adds:
- --prot_seq : adds protein seqs,
- --prg=gth : this and the following param are for adding proteins of shorter evolutionary distance
- --gth2traingenes
braker.pl --genome="$genome" --species="$species" --prot_seq="$protein" --epmode
--softmasking --gff3
Following this, we run TSEBRA
echo `hostname`
export LC_ALL=en_US.UTF-8
export LANG=en_US.UTF-8
export LANGUAGE=en_US.UTF-8
module load python/3.6.3
tsebra=/home/FCAM/vvuruputoor/TSEBRA_1.0.3/bin
b1=/core/labs/Wegrzyn/annotationtool/testSpecies/PlantSet/arabidopsis/analysis/annotation/braker/braker/augustus.hints.gtf
b2=/core/labs/Wegrzyn/annotationtool/testSpecies/PlantSet/BRAKER+TSEBRA/arabidopsis/braker2_v2.1.6/braker/augustus.hints.gtf
config=/home/FCAM/vvuruputoor/TSEBRA_1.0.3/config/default.cfg
h1=/core/labs/Wegrzyn/annotationtool/testSpecies/PlantSet/arabidopsis/analysis/annotation/braker/braker/hintsfile.gff
h2=/core/labs/Wegrzyn/annotationtool/testSpecies/PlantSet/BRAKER+TSEBRA/arabidopsis/braker2_v2.1.6/braker/hintsfile.gff
$tsebra/tsebra.py -g $b1,$b2 -c $config -e $h1,$h2 -o braker1+2_combined.gtf
3. Long Read -
- protein from species of interest - derived from long-read data
- use the"--trainFromGth" flag instead of "--gth2traingenes"
braker.pl --species=yourSpecies_long --genome=genome.fasta \
--prot_seq=longReadSpecies.fa --prg=gth --trainFromGth \
--softmasking 1 \
--gff3 \
--cores 10
4. Long and Short Read-
- combining short and long-read data for BRAKER annotation
Command
new command name
5. LTR masking + BRAKER-
- this uses the extra LTR masking as well as the Run 1 script
Runs 1, 3, 4, and 5 are also run with Stringtie prepared de novo proteins
Genome annotation with MAKER
Safety information
The genome for this step is the softmasked genome from RepeatModeler2, with the LTRStruct flag ON.
MAKER is run in iterations. The first step is to fill in the control files for MAKER.
Round 1: Input data includes genome, EST evidences (preferably externally aligned evidences through exonerate). The script to run MAKER is the same through all rounds:
Command
The most important step is to fill in the control files before running this step.
MAKER
The next step is to run AUGUSTUS and SNAP
Command
AUGUSTUS
Command
SNAP
gFACs
- Run transcriptome and protein alignments through gFACs - use gfacs_aln.sh (new script)
- gfacs_aln.sh calls gFACs.pl
- gmap and genomeThreader output is used as input
- Options
- -f gmap_2017_03_17_gff3 or genomethreader_1.6.6_gff3
- --splice-rescue
- --statistics
- --statistics-at-every-step
- --splice-table
- --min-exon-size 3
- --min-intron-size 3
- --get-fasta-without-introns
- --unique-genes-only
- --create-gtf
- --get-protein-fasta
- The last option creates protein FASTA which should run through BUSCO
2. Run the output of all four braker runs through gFACs, both filtered and unfiltered
2a. Filtered
- -f braker_2.1.5_gff3
- --splice-rescue
- --statistics
- --statistics-at-every-step
- --splice-table
- --get-fasta-without-introns
- --get-fasta-with-introns
- --min-intron-size 10
- --min-exon-size 10
- --mins-CDS-size 300
- --rem-start-introns
- --rem-end-introns
- --get-protein-fasta
- --create-gtf
2b. Unfiltered
- --splice-rescue
- --statistics
- --statistics-at-every-step
- --splice-table
- --create-gtf
ENTAP
- database information at https://bioinformatics.uconn.edu/databases/
- Run EnTAP - entap.sh
- can be run with BAM/SAM file or braker output (augustus.hints.aa)
- BAM file uses flags --ontology 0, -a <BAMFILE>
- contaminants specified by -c flags, should contaminants be specific to species of interest?
- see https://entap.readthedocs.io/en/latest/basic_usage.html#id3 for additional flags
- should have multipule .dmnd files (at least 2, refseq and uniprot)